
Биоинформа́тика — междисциплинарная область, объединяющая общую биологию, молекулярную биологию, кибернетику, генетику, химию, компьютерные науки, математику и статистику. Крупномасштабные биологические проблемы, требующие анализа больших объёмов данных, решаются биоинформатикой с вычислительной точки зрения. Биоинформатика главным образом включает в себя изучение и разработку компьютерных методов и направлена на получение, анализ, хранение, организацию и визуализацию биологических данных.

Однонуклеотидный полиморфизм — отличия последовательности ДНК размером в один нуклеотид в геноме представителей одного вида или между гомологичными участками гомологичных хромосом. Применяется в качестве генетических ма́ркеров для изучения неравновесного сцепления локусов и полногеномного поиска ассоциаций.
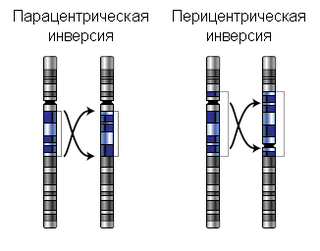
Инве́рсия — хромосомная перестройка, при которой происходит поворот участка хромосомы на 180°. Инверсии являются сбалансированными внутрихромосомными перестройками. Различают парацентрические и перицентрические инверсии. Инверсии играют роль в эволюционном процессе, видообразовании и в нарушениях фертильности.
Полиморфизм длин рестрикционных фрагментов — способ исследования геномной ДНК путём разрезания ДНК с помощью эндонуклеаз рестрикции и дальнейшего анализа размеров образующихся фрагментов (рестриктов) путём гель-электрофореза.

Проект «Геном человека» — завершённый международный научно-исследовательский проект, главной целью которого было определение последовательности пар оснований, которые составляют ДНК человека, а также выявление, картирование и секвенирование всех генов человеческого генома как с физической, так и с функциональной точки зрения. Этот проект остается крупнейшим международным биологическим проектом, когда-либо проводившимся в биологии. К 2003 году было секвенировано лишь 85 % генома человека, проект был завершён в 2022 году, когда было достигнуто полное секвенирование генома человека.
Вариация числа копий — вид генетического полиморфизма, к которому относят различия индивидуальных геномов по числу копий хромосомных сегментов размером от 1 тыс. до нескольких млн. пар оснований. CNV возникают в результате несбалансированных хромосомных перестроек, таких как делеции и дупликации. Значительный полиморфизм по CNV у человека стал очевиден после окончания полного секвенирования нескольких геномов. Крупные делеции или дупликации могут быть выявлены при микроскопическом анализе метафазных хромосом, однако подавляющая часть CNV выявляется при помощи сравнительной геномной гибридизации и при полногеномном SNP-генотипировании.
Бисульфи́тное секвени́рование — общее название группы методов, направленных на изучение паттерна метилирования ДНК посредством обработки её бисульфитом.

Са́узерн-блот, Са́узерн-бло́ттинг, бло́ттинг по Са́узерну, блот Са́зерна, бло́ттинг Са́зерна, са́зерн-блот, са́зерн-бло́ттинг — метод, применяемый в молекулярной биологии для выявления определённой последовательности ДНК в образце. Он заключается в переносе разделённых электрофорезом в агарозном геле фрагментов ДНК на мембранный фильтр и последующем обнаружении в них известной последовательности из ДНК-зонда с помощью гибридизации с ним. Метод называется по имени изобретателя, английского биолога Эдвина Саузерна.

Метагено́мика — раздел молекулярной генетики, в котором изучается генетический материал, полученный из образцов окружающей среды. Метагеномика изучает набор генов всех микроорганизмов, находящихся в образце среды, — метагеном. Метагеномный анализ позволяет определить видовое разнообразие исследуемого образца без необходимости выделения и культивирования микроорганизмов.
Международная организация по изучению генома человека — организация, созданная в рамках проекта «Геном человека». HUGO была создана в 1989 году в качестве международной организации, прежде всего для стимулирования сотрудничества между генетиками во всем мире. С момента образования в HUGO вошли 220 учёных из разных стран, в том числе пять советских биологов. С 2013 года президентом организации является греческий генетик Стилианос Антонаракис.
Секвенирование нового поколения — группа методов определения нуклеотидной последовательности ДНК и РНК для получения формального описания её первичной структуры. Технология методов секвенирования нового поколения позволяет «прочитать» единовременно сразу несколько участков генома, что является главным отличием от более ранних методов секвенирования. NGS осуществляется с помощью повторяющихся циклов удлинения цепи, индуцированного полимеразой, или многократного лигирования олигонуклеотидов. В ходе NGS могут генерироваться до сотен мегабаз и гигабаз нуклеотидных последовательностей за один рабочий цикл.
Персональная геномика является разделом геномики, связанным с секвенированием и анализом генома человека. Стадия генотипирования использует различные методы, включая однонуклеотидно полиморфные (МНП) анализирующие чипы, а также частичное или полное секвенирование генома. После расшифровки генотипа его можно проанализировать при помощи опубликованной литературы для определения вероятности риска заболеваний.

Секвени́рование экзо́ма — секвенирование всех белок-кодирующих генов в геноме. Под секвенированием экзома подразумеваются две операции: во-первых, отбор экзонов. В зависимости от организма экзоны покрывают 1—2 % генома. У человека их насчитывается около 180 000, примерно 1 % от всего генома, или приблизительно 30 миллионов пар оснований. Во-вторых, секвенирование экзонов с использованием любой платформы высокопроизводительного секвенирования ДНК и анализ полученных результатов.

Секвенирование древней ДНК — определение нуклеотидной последовательности применительно к молекулам ДНК, извлечённым из древних биологических образцов, таких как палеонтологические и археологические находки, мумифицированные останки, засохшие остатки растений, копролиты. Анализ нуклеотидных последовательностей, полученных секвенированием древней ДНК, позволяют установить филогенетические отношения между видами и проверять гипотезы о связи изменений в окружающей среде и эволюционных изменений популяций, а также предоставляют информацию для калибровки молекулярных часов.
Визуализа́ция да́нных секвени́рования РНК — способ визуального представления данных, полученных с помощью РНК-секвенирования (RNA-seq) в наглядной форме, с помощью которого можно увидеть картирование полученных чтений на геном и анализировать уровень экспрессии гена. Существует множество программ, позволяющих осуществить визуализацию.
Атлас ракового генома или АРГ — проект, целью которого является систематизация данных о генетических мутациях, приводящих к возникновению рака. Систематизация проводится с помощью секвенирования и методов биоинформатики. Данный проект — совместная работа Национального Института Рака и Института Исследований Генома Человека, США.
BWA — программный пакет для картирования коротких прочтений на большие референсные геномы, написанный китайским биоинформатиком Хенг Ли и англичанином Ричардом Дурбиным. Является одним из широкоиспользуемых алгоритмов выравнивания, а также рекомендуется для анализа данных производителями Illumina. BWA состоит из трёх основных алгоритмов: BWA-BackTrack, BWA-SW и BWA-MEM. В основе алгоритмов BWA лежит преобразование Барроуза—Уилера, суффиксные массивы и алгоритм выравнивания Смита—Ватермана. Программный пакет умеет работать с длинными последовательностями на порядок быстрее, чем MAQ при достижении аналогичной точности выравнивания.

Секвени́рование ДНК одино́чных кле́ток — подход, позволяющий получить данные о последовательности ДНК отдельной клетки с помощью секвенирования и, следовательно, выявлять различия между отдельными клетками одноклеточных организмов, органов, тканей и клеточных субпопуляций многоклеточных организмов. Подход позволяет анализировать функциональные особенности клетки в контексте микроокружения. Секвенирование генома единичной клетки включает несколько шагов: выделение одной клетки, полногеномная амплификация, создание библиотек и секвенирование ДНК с использованием методов секвенирования нового поколения.
Международный проект HapMap является организацией, деятельность которой направлена на разработку карты гаплотипа генома человека, чтобы описать общие закономерности наследственной генетической изменчивости людей. HapMap используется, чтобы найти генетические варианты, влияющие на здоровье, болезнь и реакции на лекарственные препараты и факторы окружающей среды. Информация, получаемая в рамках проекта для проведения исследований находится в свободном доступе.
«1000 геномов» — международный проект, в котором, как было объявлено в январе 2008 года, к концу 2011 года планировалось упорядочить геномы примерно 2500 человек, чтобы создать подробный каталог генетических вариаций человека, включая однонуклеотидные полиморфизмы, индели и структурные вариации, такие как вариации числа копий генов. Это было бы безусловно самым полным исследованием геномов человека на сегодняшний день. Для реализации этого проекта институты из многих стран работали вместе, в том числе в США, Англии, Китайской Народной Республике и Германии. Полная база данных предоставляется учёным во всём мире бесплатно и является обогащением для всех областей естественных наук.