SOLiD
SOLiD (англ. Sequencing by Oligonucleotide Ligation and Detection) — технология нового поколения секвенирования ДНК, развиваемая компанией Life Technologies (https://web.archive.org/web/20080516181322/http://solid.appliedbiosystems.com/), коммерчески доступна с 2006 года. SOLiD позволяет секвенировать разом сотни миллионов и даже миллиарды коротких последовательностей.
SOLiD использует метод лигирования в отличие от других платформ, таких как Roche-454 пиросеквенирование (метод появился в 2005 году, в 2009 году уже создавал миллионы последовательностей длиной 200—400 пар оснований) и система Solexa (сейчас принадлежит Illumina) (метод появился в 2006 году, в 2009 году уже создавал миллионы последовательностей длиной 50-100 пар оснований), которые используют секвенирование с помощью синтеза.
Эти все методы снизили стоимость секвенирования в 2006 году с $0.01/основание до, примерно, $0.0001/основание и увеличили мощность с 1 млн оснований/секвенатор/день (2004 год) до более 5 млн оснований/секвенатор/день (2009 год). Существуют более 30 публикаций, описывающих использование метода в нуклеосомном позиционировании[1], транскрипционном профайлинге или цепь-специфичном RNA-Seq[2], транскрипционном профайлинге в единичной клетке[3] и пересеквенировании человеческого генома[4].
Основные черты
Общие этапы, характерные для большинства методов секвенирования нового поколения:
- Фрагментация геномной ДНК случайным образом.
- Иммобилизация одноцепочечных фрагментов на твердые бусины или на плоскую твердую поверхность.
- Амплификация фрагментов ДНК на твердой подложке с помощью ПЦР.[5]
- Секвенирование и последующая обработка каждого цикла in situ с помощью сканирования флуоресценции, хемилюминесценции и других методов.[6]
Принцип метода
Процесс секвенирования включает следующие шаги[7]:
Подготовка библиотеки
Геномная ДНК разрезается на малые фрагменты. Затем к концам каждого фрагмента пришиваются две различные нуклеотидные последовательности — адаптеры A1 и A2. В итоге в библиотеке оказываются одноцепочечные нуклеотидные последовательности A1-(фрагмент ДНК)-A2.
ПЦР в эмульсии
ПЦР ДНК-фрагментов библиотеки проводят в водных каплях в масляно-водяной эмульсии. В каждой капле — 1μм бусина, к которой присоединены одноцепочечные нуклеотидные последовательности одного из двух праймеров (P1 или P2). Эти праймеры (P1 и P2) комплементарны адаптерам A1 и A2 соответственно. В такую каплю масла добавляют последовательность из библиотеки, и праймер на бусине будет образовывать дуплекс с одним из её адаптеров. Если на бусине — праймер P1, то с ним образует дуплекс адаптер A1. Это называется отжиг праймера. После отжига праймера добавляют ДНК-полимеразу, которая достраивает вторую цепь ДНК. После проведения ПЦР и диссоциации ДНК-дуплексов на бусинах остаются одноцепочечные последовательности ДНК.
Насыщение бусин с целевой ДНК
На практике, только 30 % бусин несут целевую ДНК (ДНК-фрагмент библиотеки). Для увеличения количества таких бусин добавляют в раствор другие большие полистироловые бусины, с присоединенными одноцепочечными последовательностями другого адаптера (A2), комплементарного ПЦР-праймеру (P2). Так, каждая бусина с целевой ДНК будет составлять пару с большой бусиной за счет образования дуплекса: адаптера A2 на большой бусине и участка, соответствующего P2, бусины с целевой ДНК. Такой комплекс в итоге отделяют от «пустых» бусин, а затем его плавят, чтобы прошла диссоциация бусин с целевой ДНК и полистироловой. Эта процедура позволяет увеличить количество бусин, несущих целевую ДНК до 80 %.
Затем 3’-концы, несущие последовательность P2, модифицируют для обеспечения ковалентного связывания, необходимого на следующей стадии.
Перенос бусин
Продукты, полученные на предыдущей стадии, переносят на стеклянные пластины. Бусины иммобилизуют на стекле в случайном порядке, присоединяя их ковалентно к стеклу через модификации 3’-концов на бусинах.
Секвенирование
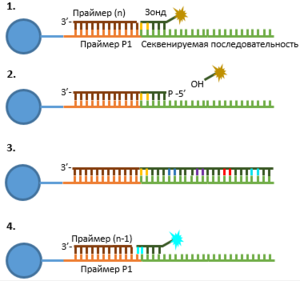
(1) Начало 1 раунда: добавление праймера длины n и 8ми нуклеотидного зонда, лигирование их друг с другом, детекция флуоресценции.
(2) Разрезание зонда, освобождение от метки.
(3) 5 последовательных циклов 1 раунда (повтор стадий (1) и (2)).
(4) Начало 2 раунда: добавление праймера длины n-1 и 8ми нуклеотидного зонда, лигирование их друг с другом, детекция флуоресценции.
Секвенирование проходит с помощью лигирования восьми-нуклеотидных зондов, меченных на 5’-конце одним из четырех различных флуорофоров. Последовательность зондов несет сайт гидролиза, находящийся между пятым и шестым нуклеотидами. Первые два основания (считая с 3’-конца) комплементарны двум нуклеотидам секвенируемой последовательности. С третьего по пятый основания зонда могут гибридизоваться с любыми тремя нуклеотидами секвенируемой ДНК. 6-8 основания зонда также могут гибридизоваться с любой последовательностью, однако они вместе с флуоресцентным красителем отщепляются от зонда в ходе реакции. Отщепление флуоресцентной метки вместе с основаниями 6-8 происходит таким образом, что на 5'-конце зонда остается фосфатная группа, способствующая следующему циклу лигирования зонда. Так, два основания каждого зонда точно комплементарны основаниям секвенируемой последовательности в позициях n+1 и n+2, n+6 и n+7 и т. д.
Процесс секвенирования состоит из пяти раундов, каждый раунд состоит из 6-7 циклов. Каждый раунд начинается с добавления универсального праймера длины n, комплементарного P1. В каждом цикле 8-ми нуклеотидные зонды добавляются и лигируются к праймеру, их первые два нуклеотида комплементарны двум нуклеотидам секвенируемой последовательности. Затем отмывают от остающихся несвязанных зондов, измеряют флуоресценцию лигированного зонда и разрезают его между пятым и шестым основаниями. По окончании последнего цикла проводят диссоциацию синтезированной цепи ДНК от матрицы, прикрепленной к бусине. Это необходимо для того, чтобы в следующем раунде уже использовать новые праймер и зонды. Праймер теперь берут длины n-1. Итак, в ходе пяти раундов используются праймеры, комплементарные P1, длины n, n-1, n-2, n-3, n-4 относительно 3'-конца P1. Таким образом можно секвенировать примерно 25 нуклеотидов последовательности.
Расшифровка данных

На выходе мы получаем данные по флуоресценции. Пространство цветов и пространство нуклеотидов содержат по 4 элемента. Каждый цвет кодирует собой 4 из 16 возможных динуклеотидов. Например, «синий» цвет флуоресцентной метки соответствует паре одинаковых нуклеотидов (то есть AA,GG,TT или CC). Дизайн матрицы преобразований цвета способствует коррекции ошибок.
Одним цветом кодируется:
- пара нуклеотидов и она же в обратном порядке (например, CA и AC)
- пара нуклеотидов и комплементарная ей пара (например, CA и GT)
- пара нуклеотидов и обратно комплементарная ей пара (например, CA и TG)
Последовательность нуклеотидов может быть единственным образом преобразована в последовательность цветов. Но последовательность цветов может быть преобразована в последовательность нуклеотидов 4 разными способами. Это похоже на взаимосоответствие между нуклеотидами и аминокислотами(цвета).
Для расшифровки последовательности нуклеотидов по цветам необходимо знать один нуклеотид.
Преимущества метода
В данном методе каждый нуклеотид прочитывается дважды, что повышает точность секвенирования. Соответственно чтобы допустить ошибку секвенирования (пропустить SNP) необходимо оба раза неправильно определить цвет флуоресцентной метки при секвенировании соседних нуклеотидов.
См. также
Примечания
- ↑ Valouev A., Ichikawa J., Tonthat T., et al. A high-resolution, nucleosome position map of C. elegans reveals a lack of universal sequence-dictated positioning (англ.) // Genome Research : journal. — 2008. — July (vol. 18, no. 7). — P. 1051—1063. — doi:10.1101/gr.076463.108. — PMID 18477713. — PMC 2493394.
- ↑ Cloonan N., Forrest A. R., Kolle G., et al. Stem cell transcriptome profiling via massive-scale mRNA sequencing (итал.) // Nature Methods : diario. — 2008. — Luglio (v. 5, n. 7). — P. 613—619. — doi:10.1038/nmeth.1223. — PMID 18516046.
- ↑ Tang F., Barbacioru C., Wang Y., et al. mRNA-Seq whole-transcriptome analysis of a single cell (англ.) // Nature Methods : journal. — 2009. — May (vol. 6, no. 5). — P. 377—382. — doi:10.1038/nmeth.1315. — PMID 19349980.
- ↑ McKernan K. J., Peckham H. E., Costa G. L., et al. Sequence and structural variation in a human genome uncovered by short-read, massively parallel ligation sequencing using two-base encoding (англ.) // Genome Research : journal. — 2009. — September (vol. 19, no. 9). — P. 1527—1541. — doi:10.1101/gr.091868.109. — PMID 19546169. — PMC 2752135.
- ↑ Chetverin, NAR, 1993, Vol.21, No. 10 2349—2353
- ↑ MATTHEW E. HUDSON (2008) Sequencing breakthroughs for genomic ecology and evolutionary biology. Molecular Ecology Resources 8 (1) , 3-17
- ↑ Applied Biosystems. Дата обращения: 12 июля 2019. Архивировано из оригинала 8 сентября 2013 года.