
Аути́зм — расстройство, возникающее вследствие нарушения развития головного мозга и характеризующееся выраженным и всесторонним дефицитом социального взаимодействия и общения, а также ограниченными интересами и повторяющимися действиями. Все указанные признаки начинают проявляться в возрасте до трёх лет. Схожие состояния, при которых отмечаются более мягкие признаки и симптомы, относят к расстройствам аутистического спектра.

Болезнь Гентингтона — аутосомно-доминантное генетическое заболевание нервной системы, характеризующееся постепенным началом обычно в возрасте 30—50 лет и сочетанием прогрессирующего хореического гиперкинеза и психических расстройств. Заболевание вызывается умножением кодона CAG в гене HTT. Этот ген кодирует 350-kDa белок хантингтин с неизвестной функцией. В гене дикого типа у разных людей присутствует разное количество CAG-повторов, однако, когда число повторов превышает 36, развивается болезнь. Нейроморфологическая картина характеризуется атрофией стриатумa, а на поздней стадии также атрофией коры головного мозга.
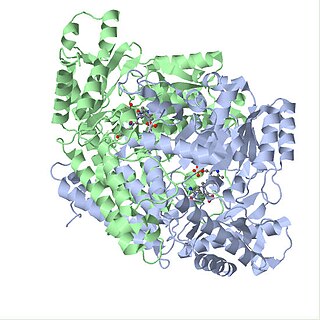
Глутаматдекарбоксилаза (англ. Glutamate decarboxylase, GAD, GAD65, GAD67, шифр КФ 4.1.1.15) — фермент, катализирующий преобразование глутамата в ГАМК посредством декарбоксилирования по следующей реакции:
- HOOC-CH2-CH2-CH(NH2)-COOH → CO2 + HOOC-CH2-CH2-CH2NH2.
DISC1 или Disrupted In Schizophrenia 1 — белок, кодируемый одноимённым геном. Мутации, нарушающие функционирование белка, связаны как с шизофренией, так и с другими психическими расстройствами. При их воспроизведении в животных моделях обнаруживаются типичные для шизофрении отклонения. Нарушение молекулярных взаимодействий, в которых участвует DISC1, может играть роль в развитии шизофрении, биполярного расстройства и депрессии. Исследования сцепленного наследования и полиморфизмов противоречивы и говорят о незначительном вкладе функциональных вариантов гена в общий риск развития шизофрении.

Адаптерный белок синтазы оксида азота-1 (нейрональной) — цитозольный белок, связывающий сигнальную молекулу nNOS. На C-окончании содержит PDZ-связывающий домен, взаимодействующий с nNOS, а на N-окончании — PTB-домен, взаимодействующий с малым мономерным G-белком DEXRAS1. Исследования на животных показывают, что NOS1AP является адаптерным белком, связывающим nNOS с некоторыми другими белками, такими как DEXRAS1 и синапсины.

TGFBI — белок человека. Экспрессия TGFBI индуцируется фактором роста TGF-beta. Альтернативное название кератоэпителин свидетельствует об узко выраженной картине экспрессии белка в тканях глаза: он производится клетками эпителия роговицы и стромальными кератоцитами. Ген был впервые описан в 1997 году, изначально получив название BIGH3.

Синдром MELAS — прогрессирующее нейродегенеративное заболевание, характеризующееся проявлениями, перечисленными в названии, и сопровождается полиморфной симптоматикой — диабетом, судорогами, снижением слуха, сердечными заболеваниями, низким ростом, эндокринопатиями, непереносимостью физических нагрузок и нейропсихиатрическими отклонениями. В каждом конкретном случае набор симптомов и их тяжесть может сильно отличаться, поскольку синдром связан с мутациями во многих генах: MTTL1, MTTQ, MTTH, MTTK, MTTS1, MTND1, MTND5, MTND6, MTTS2. Мутации могут возникать впервые у конкретного пациента, либо наследоваться по материнской линии. Всего к 2009 году было обнаружено 23 миссенсных точечных мутаций и 4 делеции мтДНК, приводящих к MELAS, однако продолжаются сообщения о новых пациентах с симптомами расстройства при отсутствии известных мутаций.
TNFRSF13C, или рецептор фактора, активирующего B-лимфоциты — мембранный белок, рецептор из надсемейства рецепторов факторов некроза опухоли, играет важную роль в регуляции иммунного ответа. Продукт гена человека TNFRSF13C.
Кристаллическая дистрофия Bietti (BCD), также называемая кристаллическая корнеоретинальная дистрофия Bietti , является редким аутосомно — рецессивным заболеванием глаз и названа в честь доктора G.B. Bietti.
Геномная нестабильность определяется высокой частотой мутаций в геноме клеточной линии. Эти мутации могут включать в себя изменения в последовательности нуклеиновых кислот, хромосомные перестройки или анеуплоидию. Геномная нестабильность является центральным фактором канцерогенеза, но также фактором некоторых нейродегенеративных заболеваний, таких как боковой амиотрофический склероз или нервно-мышечное заболевание миотоническая дистрофия.

Церебральная фолатная недостаточность — синдром, при котором в спинномозговой жидкости пациента снижено содержание 5-метилтетрагидрофолата (5-MTHF), несмотря на нормальное содержание 5-MTHF в сыворотке крови. Набор симптомов варьирует в зависимости от возраста начала заболевания и его причины, и может включать дискинезию, атаксию, эпилептические приступы, задержку психомоторного развития.
Синдром Смит — Магенис (СМС) — наследственное заболевание, имеет такие проявления, как умственная отсталость, аномалии лица, проблемы со сном и многочисленные поведенческие проблемы, в том числе и такие, как самоповреждение. Частота синдрома Смит — Магенис составляет 1 случай на 15 000-25 000 человек. Синдром Смит — Магенис, как правило, обусловлен микроделецией в коротком плече 17-ой хромосомы (17p), и иногда его называют 17p-синдром.
Недостаточность дигидроптеридинредуктазы — генетическое расстройство синтеза тетрагидробиоптерина (BH4), вызываемое мутациями гена QDPR. Мутации гена нарушают работу фермента 6,7-дигидроптеридинредуктазы (ДГПР), отвечающего за регенерацию BH4. Тип наследования - аутосомно-рецессивный.
Недостаточность тетрагидробиоптерина – редкое состояние, при котором пациент страдает от недостатка тетрагидробиоптерина - важного кофермента, необходимого для синтеза моноаминовых нейромедиаторов дофамина и серотонина. Патогенез этого состояния разнообразен: на начало 2021 года было известно шесть редких нейрометаболических заболеваний, нарушающих процесс биосинтеза либо рециклинга BH4. При четырех из этих шести заболеваний у пациента может обнаруживаться повышение концентрации фенилаланина. Терапия в основном заключается в восполнении уровней нейромедиаторов и в коррекции гиперфенилаланинемии.

Капикуа — белок человека, способный подавлять экспрессию ряда генов. Капикуа кодируется геном CIC, расположенным на длинном плече 19 хромосомы.
DDX3X-синдром — генетическое заболевание, развивающееся преимущественно у лиц женского пола. У пациентов наблюдается задержка умственного развития, признаки аутизма, СДВГ, сниженный мышечный тонус. Причина развития синдрома — мутации, затрагивающие ген DDX3X, расположенный на X-хромосоме. Клиническая картина зависит от характера конкретной мутации.
Пиридоксин-зависимая эпилепсия - тяжелая форма эпилепсии, при которой приступы с трудом поддаются контролю обычными противоэпилептическими препаратами. Заболевание развивается из-за мутаций гена ALDH7A1, приводящих к ускоренной деактивации активной формы витамина B6 в организме. Как правило, приступы начинаются вскоре после рождения, реже - через несколько месяцев, крайне редко - через несколько лет. Раннее назначение больших доз витамина B6 позволяет купировать приступы.
Аутосомно-доминантная недостаточность ГТФ-циклогидролазы 1 — заболевание, вызываемое нарушением работы фермента ГТФ-циклогидролаза 1, играющего важную роль в цепочке синтеза тетрагидробиоптерина. Это состояние является одной из нескольких известных причин недостаточности тетрагидробиоптерина, а также самой распространённой причиной дофа-зависимой дистонии. У пациентов наблюдается генерализованная дистония с изменением тяжести симптомов в течение дня, а назначение леводопы приводит к радикальному улучшению состояния.
Слабовыраженная гиперфенилаланинемия без дефицита тетрагидробиоптерина — редкое метаболическое заболевание, при котором наблюдается небольшое повышение концентрации фенилаланина в крови, а также ряд неврологических нарушений различного характера и степени тяжести, в том числе двигательные нарушения и расстройства интеллекта. Заболевание наследуется по аутосомно-рецессивному типу. Альтернативное название заболевания — недостаточность DNAJC12.
Синдром гиперфосфатазии с умственной отсталостью – заболевание, при котором у пациентов отмечается умственная отсталость различной степени тяжести и устойчивое повышение активности щелочной фосфатазы в крови. По состоянию на 2020 год было известно шесть генов, мутации которых приводят к развитию синдрома Мабри.