
Насле́дственные заболева́ния — заболевания, возникновение и развитие которых связано с различными дефектами и нарушениями в наследственном аппарате клеток. В основе наследственных заболеваний лежат мутации: хромосомные, генные и митохондриальные. Наследственные заболевания могут быть обусловлены мутациями, передаваемыми в семьях по наследству, или мутациями, вновь возникшими в клетках зародышевой линии, в зиготе или на очень ранних этапах развития. Наследственные болезни многочисленны и разнообразны по проявлениям.

Синдром Герстмана — Штраусслера — Шейнкера — очень редкое, обычно семейное, смертельное нейродегенеративное заболевание, поражающее пациентов в возрасте от 20 до 60 лет. Классифицируется как трансмиссивная спонгиоформная энцефалопатия, причиной заболевания является мутация гена прионового белка. Впервые было описано австрийскими неврологами Йозефом Герстманом, Эрнстом Штраусслером и Ильёй Шейнкером в 1936 году.
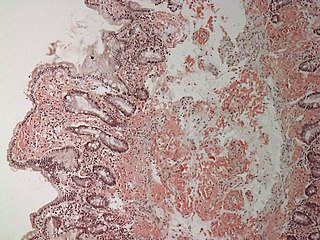
Амилоидóз — нарушение белкового обмена, сопровождающееся образованием и отложением в тканях специфического белково-полисахаридного комплекса — амилоида.
Перницио́зная анеми́я (от лат. perniciosus — гибельный, опасный), или B12-дефицитная анемия, или мегалобластная анемия, или болезнь Аддисона — Бирмера, или (устаревшее название) злока́чественное малокро́вие, — заболевание, обусловленное нарушением кроветворения, причиной которого может быть несколько факторов, основным из которых является недостаток в организме витамина B12.

Боле́знь Тея — Са́кса (GM2 ганглиозидоз, ранняя детская амавротическая идиотия) — редкое наследственное заболевание с аутосомно-рецессивным типом наследования, поражающее центральную нервную систему (спинной и головной мозг, а также менингеальные оболочки). Относится к группе лизосомных болезней накопления. Названо в честь британского офтальмолога Уоррена Тея (англ. Warren Tay, 1843—1928) и американского невролога Бернарда Сакса (англ. Bernard Sachs, 1858—1944), которые впервые описали это заболевание независимо друг от друга в 1881 и 1887 годах соответственно.
Ацерулоплазминемия — редкое заболевание, вызванное мутациями гена, кодирующего синтез церулоплазмина. Данный белок, основная функция которого заключается в транспорте меди, также участвует в метаболизме железа, и мутации, нарушающие эту функцию церулоплазмина, приводят к токсичным отложениям железа в мозге, печени, поджелудочной железе.

Врождённая гиперплазия коры надпочечников (ВГКН) — группа заболеваний, наследуемых по аутосомно-рецессивному пути, при которых нарушается выработка кортизола надпочечниками. Гены, связанные с гиперплазией надпочечников, кодируют ферменты, участвующие в стероидогенезе — цепочке реакций по преобразованию холестерина в стероиды.

Поликисто́з почек представляет собой генетическое заболевание, проявляющееся кистозным перерождением паренхимы почек. Одна из форм поликистозной дисплазии почек. Болезнь затрагивает не только сами почки, но часто также другие органы (печень).

Спиноцеребеллярная атаксия — прогрессирующее нейродегенеративное наследственное заболевание различных типов, каждый из которых рассматривается как отдельная болезнь. На сегодняшний день известно более 20 типов этого заболевания
Генные болезни — это большая группа заболеваний, возникающих в результате повреждения ДНК на уровне гена. Термин употребляется в отношении моногенных заболеваний, в отличие от более широкой группы — Наследственные заболевания (см.)
Не́оната́льный са́харный диабе́т — редко встречающееся гетерогенное по этиологии заболевание, проявляющееся в первые 6 месяцев жизни. Различают две основные клинические группы:
- транзито́рный (преходящий) неонатальный сахарный диабет и
- пермане́нтный (персистирующий) неонатальный сахарный диабет.

DIDMOAD-синдро́м — аутосомно-рецессивно наследуемый синдром, ассоциированный с инсулинозависимым сахарным диабетом и прогрессирующей атрофией диска зрительного нерва, которые выявляют до 16-летнего возраста. Сочетается с двусторонней прогрессирующей нейросенсорной тугоухостью, несахарным диабетом центрального генеза, дисфункцией автономной нервной системы, приводящей к развитию нейропатического мочевого пузыря и другим проявлениям нейродегенерации, включающими мозжечковую атаксию, миоклональную эпилепсию и атрофию ствола головного мозга. Развёрнутая клиническая картина (фенотипически) встречается приблизительно у 75 % пациентов. Сахарный диабет неаутоиммунного генеза, клинические проявления недостаточности инсулина проявляются приблизительно в 6-летнем возрасте. Средняя продолжительность жизни достигает 30 лет, в течение этого срока происходит развитие полного фенотипа данного синдрома.
Болезнь (синдром) Рефсума — редкое наследственное заболевание, которое проявляется в том, что вследствие дефекта выработки специального фермента α-окисления в организме человека накапливается фитановая кислота. Её концентрация повышается в клетках центральной и периферической нервных систем, а также в тканях внутренних органов. Через определенное время содержание этой кислоты в миелине вырастает до половины от общего количества жирных кислот в нём, что приводит к перекисному окислению липидов и деструкции миелиновой оболочки.
Втори́чные фо́рмы са́харного диабе́та — разнородная группа заболеваний, к которой относится сахарный диабет, встречающийся на фоне другой клинической патологии, которая может и не сочетаться с сахарным диабетом. Для большинства заболеваний из этой группы этиологические факторы раскрыты. Кроме того, к этой группе заболеваний также относят и некоторые генетические (наследственные) синдромы, в том числе аномалии инсулиновых рецепторов. При вторичных формах сахарного диабета отсутствуют ассоциации с HLA-антигенами, данные за аутоиммунное поражение и антитела к островковой ткани поджелудочной железы.

Железосерные кластеры — элементоорганические соединения, группа белковых кофакторов, обладающих окислительно-восстановительным (Red/Ox) потенциалом в районе от −500 мВ до +300 мВ. Red/Ox-потенциал зависит от структуры и конформации белка, что делает эти кофакторы важнейшими участниками окислительно-восстановительных реакций в клетке. Железосерные кластеры способны принимать или отдавать электроны. Белки, содержащие железосерные кластеры, являются эволюционно древними и распространены во всех царствах, включая животных, растения, грибы, бактерии и археи. Мутации по генам метаболизма Fe—S-кластеров являются причиной многих тяжёлых заболеваний или летальны.
Нейродегенеративные заболевания — группа в основном медленно прогрессирующих, наследственных или приобретённых заболеваний нервной системы. Общим для этих заболеваний является прогрессирующая гибель нервных клеток (нейродегенерация), ведущая к различным неврологическим симптомам — прежде всего, к деменции и нарушению движений. Заболевания могут наступить в различном возрасте, протекают диффузно или генерализированно, гистологически определяется специфический тип изменений.
GM2-ганглиозидо́з — тяжёлое наследственное заболевание, развивающееся в результате дефицита или недостаточной активности фермента гексозаминидазы и накопления в клетках ганглиозидов. Относится к лизосомным болезням накопления и имеет три варианта, связанные с мутациями в разных генах, которые оказывают вляние на активность общей гексозаминидазы. Два варианта заболевания больше известны под индивидуальными именами, полученными в честь авторов, впервые описавших их клиническую картину: болезнь Тея — Сакса, болезнь Сандхоффа. Третий вариант этой болезни носит название GM2 ганглиозидоз, вариант АБ. Болезнь Тея — Сакса вызвана мутацией в гене HEXA, кодирующий альфа-субъединицу гексоаминидазы А. Болезнь Сандхоффа вызвана мутацией в гене HEXB, кодирующий бета-субъединицу гексоаминидаз А и Б. GM2 ганглиозидоз, АБ вариант связан с нарушением в гене GM2, кодирующий белок-активатор GM2A.

Болезнь Шарко — Мари — Тута (ШМТ), или наследственная моторно-сенсорная нейропатия (НМСН) — наследственная периферическая нейропатия с хроническим прогрессирующим течением. При этой болезни больные страдают от слабости и атрофии мышц дистальных отделов конечностей, деформации стоп и кистей, у них наблюдается снижение сухожильных рефлексов, изменение походки, потеря чувствительности в конечностях. В основе клинических проявлений болезни лежит поражение двигательных и чувствительных периферических нервных волокон. Болезнь Шарко — Мари — Тута диагностируется с приблизительной частотой 1 на 2500 человек. Первое проявление болезни чаще всего происходит в подростковом возрасте или в раннем взрослом возрасте. Тяжесть симптомов сильно различается даже среди членов одной семьи с этим заболеванием. Болезнь Шарко — Мари — Тута нередко ведёт к ограничению трудоспособности и к инвалидизации, при этом большинство пациентов имеют нормальную продолжительность жизни. Болезнь Шарко — Мари — Тута является генетически крайне неоднородным заболеванием, симптомы этой болезни могут быть вызваны мутациями в более чем двух десятках генов, хотя большая часть заболеваний вызвана мутациями в генах PMP22, MPZ, GJB1 и MFN2. Наследование болезни чаще всего аутосомно-доминантное, однако может быть аутосомно-рецессивным и Х-сцепленным.
Спастическая спинальная параплегия — неврологическое заболевание верхнего двигательного нейрона.
Генетическая антисипация — это явление, при котором симптомы генетических заболеваний появляются у потомков в более раннем возрасте, чем у родителя, и усиливаются в каждом последующем поколении. Антисипация чаще проявляется в нарушении тринуклеотидного повтора. Например, болезнь Гентингтона или миотоническая дистрофия, которые обусловлены динамическими мутациями ДНК. Во всех болезнях связанных с антисипацией отмечаются неврологические симптомы.
