
Фенилкетонури́я — наследственное заболевание группы ферментопатий, связанное с нарушением метаболизма аминокислот, главным образом фенилаланина. Несоблюдение низкобелковой диеты сопровождается накоплением фенилаланина и его токсических продуктов, что приводит к тяжёлому поражению ЦНС, проявляющемуся, в частности, в виде нарушения умственного развития. Одно из немногих наследственных заболеваний, поддающихся успешному лечению.
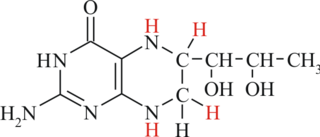
Тетрагидробиоптерин (BH4) — кофермент, участвующий в ряде важных биохимических реакций, в частности в процессах гидроксилирования на этапе промежуточного обмена ароматических аминокислот.
Нейролептические экстрапирамидные расстройства — комплекс проявляющихся двигательными нарушениями неврологических осложнений, связанных с применением препаратов-нейролептиков (антипсихотиков). Термин «лекарственные экстрапирамидные расстройства» включает в себя также нарушения, вызванные приёмом других средств, изменяющих дофаминергическую активность: например, антидепрессантов, антиаритмических препаратов, холиномиметиков, лития, антипаркинсонических средств, антиконвульсантов.
Рецептор фолиевой кислоты альфа, или рецептор фолиевой кислоты 1, — высокоаффинный клеточный рецептор к фолиевой кислоте и нескольким её производным, относится к семейству белков рецепторов фолиевой кислоты. У человека описано 4 типа рецепторов из этого семейства. В клетке рецептор обеспечивает доставку в клетку 5-метилтетрагидрофолата, кофактора, необходимого для клеточной пролиферации. Вместе с рецептором фолиевой кислоты бета является мишенью для антираковой терапии, так как блокировка транспорта фолата в раковые клетки предотвращает их дальнейшее размножение.

Синдро́м Ка́рнея — редкое наследственное заболевание с аутосомно-доминантным типом наследования. Характеризуется образованием у детей множественных опухолей. Следует отличать от триады Карни.

Нейродегенерация с отложением железа в мозге или Болезнь Галлервордена — Шпатца — очень редкое нейродегенеративное заболевание, сопровождающееся отложением железа в базальных ганглиях. Это аутосомно-рецессивно наследуемое заболевание было впервые описано в 1922 году Юлиусом Галлеворденом и Гуго Шпатцем. Частота заболевания в среднем 1-3 человека на 1 миллион.
Альфа-маннозидо́з — редкое аутосомно-рецессивное наследственное заболевание из группы лизосомных болезней накопления, связанное с нарушением метаболизма олигосахаридов в результате снижения активности фермента лизосом альфа-маннозидазы. Также заболевание наносит ущерб в животноводстве.
Синдро́м Ке́рнса — Се́йра (англ. Kearns–Sayre syndrome, сокращённо KSS) — митохондриальная миопатия с типичным началом до 20-летнего возраста. KSS является более серьёзным синдромным вариантом хронической прогрессирующей внешней офтальмоплегии, синдром, который характеризуется изолированным поражением мышц, контролирующих движения век и контролирующих движения глаз. Это приводит к птозу и офтальмоплегии соответственно. KSS включает в себя триаду уже описанных CPEO, двустороннюю пигментную ретинопатию и блокаду сердца. Другие области участия могут включать в себя мозжечковую атаксию, проксимальную мышечную слабость, глухоту, сахарный диабет, дефицит гормона роста, гипопаратиреоз или другие эндокринные нарушения. В обоих этих заболеваниях вовлечение мышц может начаться односторонним, но всегда развивается в двусторонний дефицит, который является прогрессирующим.

Церебральная фолатная недостаточность — синдром, при котором в спинномозговой жидкости пациента снижено содержание 5-метилтетрагидрофолата (5-MTHF), несмотря на нормальное содержание 5-MTHF в сыворотке крови. Набор симптомов варьирует в зависимости от возраста начала заболевания и его причины, и может включать дискинезию, атаксию, эпилептические приступы, задержку психомоторного развития.

Недостаточность декарбоксилазы ароматических аминокислот - редкое аутосомно-рецессивное генетическое заболевание, вызываемое мутациями гена DDC, кодирующего соответствующий фермент.
6,7-Дигидроптеридинредуктаза — фермент, катализирующий следующую реакцию:
- 5,6,7,8-тетрагидроптеридин + NAD+ = 6,7-дигидроптеридин + H+ + NADH
Недостаточность дигидроптеридинредуктазы — генетическое расстройство синтеза тетрагидробиоптерина (BH4), вызываемое мутациями гена QDPR. Мутации гена нарушают работу фермента 6,7-дигидроптеридинредуктазы (ДГПР), отвечающего за регенерацию BH4. Тип наследования - аутосомно-рецессивный.

Недостаточность 6-пирувоилтетрагидроптеринсинтазы - редкое заболевание, наследуемое по аутосомно-рецессивному типу. Мутации гена PTS при данном заболевании нарушают одну из реакций в цепочке синтеза тетрагидробиоптерина (BH4), в результате чего у пациента развивается избыток аминокислоты фенилаланина. В свою очередь сбой производства тетрагидробиоптерина, необходимого для выработки нескольких нейромедиаторов, приводит к недостатку этих нейромедиаторов. У пациентов наблюдается целый ряд симптомов, включающий аксиальную гипотонию, задержку развития, когнитивные нарушения, эпилептические приступы, но не ограничивающийся ими. Терапия заболевания заключается в восполнении недостающих нейромедиаторов и мерах, направленных на борьбу с гиперфенилаланинемией.
Недостаточность тетрагидробиоптерина – редкое состояние, при котором пациент страдает от недостатка тетрагидробиоптерина - важного кофермента, необходимого для синтеза моноаминовых нейромедиаторов дофамина и серотонина. Патогенез этого состояния разнообразен: на начало 2021 года было известно шесть редких нейрометаболических заболеваний, нарушающих процесс биосинтеза либо рециклинга BH4. При четырех из этих шести заболеваний у пациента может обнаруживаться повышение концентрации фенилаланина. Терапия в основном заключается в восполнении уровней нейромедиаторов и в коррекции гиперфенилаланинемии.
Недостаточность дигидрофолатредуктазы — редкое расстройство фолатного метаболизма, наследуемое по аутосомно-рецессивному типу. Расстройство возникает вследствие мутаций гена DHFR, кодирующего соответствующий фермент. При заболевании у пациента может отмечаться мегалобластная анемия и церебральная фолатная недостаточность, может наблюдаться ряд неврологических симптомов различной степени тяжести. Так, пациент может страдать отставанием в развитии, у него могут развиться эпилептические приступы.
Пиридоксин-зависимая эпилепсия - тяжелая форма эпилепсии, при которой приступы с трудом поддаются контролю обычными противоэпилептическими препаратами. Заболевание развивается из-за мутаций гена ALDH7A1, приводящих к ускоренной деактивации активной формы витамина B6 в организме. Как правило, приступы начинаются вскоре после рождения, реже - через несколько месяцев, крайне редко - через несколько лет. Раннее назначение больших доз витамина B6 позволяет купировать приступы.
Аутосомно-доминантная недостаточность ГТФ-циклогидролазы 1 — заболевание, вызываемое нарушением работы фермента ГТФ-циклогидролаза 1, играющего важную роль в цепочке синтеза тетрагидробиоптерина. Это состояние является одной из нескольких известных причин недостаточности тетрагидробиоптерина, а также самой распространённой причиной дофа-зависимой дистонии. У пациентов наблюдается генерализованная дистония с изменением тяжести симптомов в течение дня, а назначение леводопы приводит к радикальному улучшению состояния.
Недостаточность сепиаптеринредуктазы — заболевание, вызываемое нарушением работы фермента сепиаптеринредуктазы, участвующего в цепочке синтеза тетрагидробиоптерина. Развивающаяся при этом заболевании недостаточность тетрагидробиоптерина вызывает снижение уровней нейромедиатора дофамина, вследствие чего у пациентов отмечаются двигательные нарушения, такие как дистония и окулогирные кризы.
Недостаточность птерин-4-альфа-карбиноламиндегидратазы — заболевание, вызываемое нарушением работы фермента птерин-4-альфа-карбиноламиндегидратаза, участвующего в цепочке синтеза тетрагидробиоптерина. По состоянию на 2020 год недостаточность птерин-4-альфа-карбиноламиндегидратазы была самым редко описываемым в медицинских журналах типом недостаточности тетрагидробиоптерина.
Слабовыраженная гиперфенилаланинемия без дефицита тетрагидробиоптерина — редкое метаболическое заболевание, при котором наблюдается небольшое повышение концентрации фенилаланина в крови, а также ряд неврологических нарушений различного характера и степени тяжести, в том числе двигательные нарушения и расстройства интеллекта. Заболевание наследуется по аутосомно-рецессивному типу. Альтернативное название заболевания — недостаточность DNAJC12.
