Лизосо́ма — окружённая мембраной клеточная органелла, в полости которой поддерживается кислая среда и находится множество растворимых гидролитических ферментов. Лизосома отвечает за внутриклеточное переваривание макромолекул, в том числе при аутофагии; лизосома способна к секреции своего содержимого во время экзоцитоза; также лизосома участвует в некоторых внутриклеточных сигнальных путях, связанных с метаболизмом и ростом клетки.
Болезнь Гирке — гликогеноз вызванная недостаточностью глюкозо-6-фосфатазы.
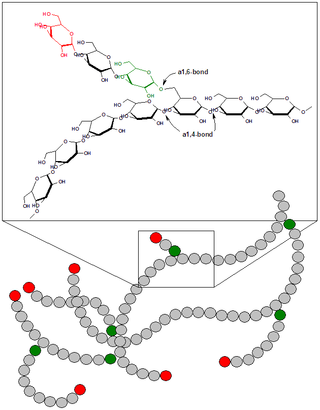
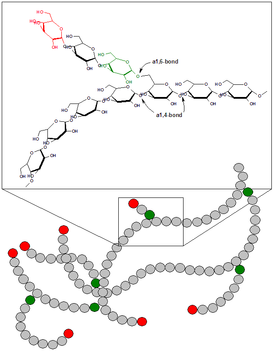
Болезнь Андерсена — гликогеноз, семейный цирроз печени, вызванный дефектом фермента амило-(1,4-1,6)-трансглюкозилазы.

Болезнь Мак-Ардля — гликогеноз, связанный с дефектом мышечной фосфорилазы.
Гликогенозы — общее название синдромов, обусловленных наследственными дефектами ферментов, участвующих в синтезе или расщеплении гликогена.

Болезнь Гоше́ — наследственное заболевание типа сфинголипидозов, является самой распространённой из лизосомных болезней накопления. Развивается в результате недостаточности фермента глюкоцереброзидазы, которая приводит к накоплению глюкоцереброзида во многих тканях, включая селезёнку, печень, почки, лёгкие, мозг и костный мозг. Заболевание связано с рецессивной мутацией в гене GBA, расположенном в 1-й хромосоме, и поражает как мужчин, так и женщин. Заболевание названо в честь французского врача Филиппа Гоше, который первым описал его в 1882.

Эдмонд Генри Фи́шер — швейцарский и американский биохимик, лауреат Нобелевской премии по физиологии или медицине 1992 года «За открытия, касающиеся обратимого белкового фосфорилирования как механизма биологической регуляции». Почётный профессор Вашингтонского университета (Сиэтл).

Ге́рти Тере́за Ко́ри, урождённая Ра́дниц — американский биохимик. Член Национальной академии наук США (1948).
Паренхимато́зные дистрофи́и — нарушения метаболизма в паренхиме органов.
Углеводный обмен, или метаболизм углеводов в организмах животных и человека. Метаболизм углеводов в организме человека состоит из следующих процессов:
- Расщепление в пищеварительном тракте поступающих с пищей поли- и дисахаридов до моносахаридов, дальнейшее всасывание моносахаридов из кишечника в кровь.
- Синтез и распад гликогена в тканях, прежде всего в печени.
- Гликолиз — распад глюкозы. Первоначально под этим термином обозначали только анаэробное брожение, которое завершается образованием молочной кислоты (лактата) или этанола и углекислого газа. В настоящее время понятие «гликолиз» используется более широко для описания распада глюкозы, проходящего через образование глюкозо-6-фосфата, фруктозо-1,6-дифосфата и пирувата как в отсутствие, так и в присутствии кислорода. В последнем случае употребляется термин «аэробный гликолиз», в отличие от «анаэробного гликолиза», завершающегося образованием молочной кислоты или лактата.
- Анаэробный путь прямого окисления глюкозы или, как его называют, пентозофосфатный путь.
- Взаимопревращение гексоз.
- Анаэробный метаболизм пирувата. Этот процесс выходит за рамки углеводного обмена, однако может рассматриваться как завершающая его стадия: окисление продукта гликолиза — пирувата.
- Глюконеогенез — образование углеводов из неуглеводных продуктов.
Болезнь Фарбера — очень редкая аутосомная рецессивная болезнь накопления лизосом. Ввиду дефицита фермента керамидазы накапливаются липиды, что приводит к нарушениям в суставах, печени, горле, центральной нервной системе и различных тканях. Керамидаза расщепляет жиры в клетках организма. В случае болезни Фарбера ген, ответственный за выработку этого фермента, изменён в результате мутации. Поэтому жиры расщепляются и накапливаются в разных частях тела. Появляются признаки этого расстройства.

Лизосо́мные боле́зни накопле́ния — общее название группы весьма редких наследственных заболеваний, вызванных нарушением функции внутриклеточных органелл лизосом. Эти одномембранные органоиды являются частью эндомембранной системы клетки и специализируются на внутриклеточном расщеплении веществ: гликогена, гликозаминогликанов, гликопротеинов и других. Лизосомные болезни накопления вызываются генетически обусловленным дефицитом ферментов лизосом, что приводит к накоплению макромолекул, являющихся субстратом этих ферментов, в различных органах и тканях организма.

Боле́знь По́мпе — редкое, наследственное заболевание с аутосомно-рецессивным механизмом, наследования,, связанное с повреждением мышечных, и нервных клеток по всему организму. Клиническая картина данной патологии, обусловлена накоплением гликогена, в лизосомах, вызванным недостаточностью лизосомного фермента — кислой α-1,4-глюкозидазы. Различают быстро прогрессирующую (классическую) и медленно прогрессирующую формы болезни Помпе. Несмотря на широкий спектр клинических проявлений, гликогеноза II типа, в основе всех форм болезни лежит, дефицит одного фермента, кодируемого геном GAA.

Синдром Сандхоффа — наследственное заболевание, вариант 0 GM2-ганглиозидоза из группы лизосомных болезней накопления — нарушение липидного обмена, вызванное наследственным дефицитом (недостаточностью) ферментов бета-гексозаминидазы А и Б.
Болезнь Вольмана — – это редкое аутосомно-рецессивное заболевание лизосомального накопления, вызванное повреждающими мутациями гена LIPA. Возраст начала заболевания и темпы его прогрессирования в значительной степени вариабельны и могут быть связаны с природой, лежащих в основе мутаций. Заболевание у пациентов грудного возраста имеет наиболее быстро прогрессирующее течение с развитием проявлений и симптомов в первые недели жизни; эти пациенты редко доживают до возраста, превышающего 6 месяцев. У детей старшего возраста и взрослых заболевание, обычно, характеризуется определенным сочетанием дислипидемии, гепатомегалии, повышением уровня трансаминаз и микровезикулярным стеатозом в биопсийном материале. У большей части пациентов наблюдается повреждение печени с исходом в фиброз, цирроз и печеночную недостаточность. Частыми изменениями являются повышение уровней холестерина липопротеинов низкой плотности и снижение уровней холестерина липопротеинов высокой плотности. Начиная с детского возраста, могут проявляться и нарушения со стороны сердечно-сосудистой системы. Учитывая, что эти клинические проявления могут наблюдаться и при других сердечно-сосудистых нарушениях, заболеваниях печени и метаболических расстройствах, неудивительно, что LAL-D часто не диагностируется в клинической практике.
Иоанн Кассианус Помпе (1901—1945) — голландский патолог. Изучал медицину в университете Утрехта. В своей публикации «Over idiopathische hypertrophie van het hart» (1932) описал заболевание, которое сейчас называют классическая (инфантильная) форма болезни Помпе или гликогеноз II типа.
Недоста́точность ки́слой фосфата́зы — редкое наследственное заболевание из группы лизосомных болезней накопления. Клиническая симптоматика дефицита кислой фосфатазы развивается в раннем возрасте. Характеризуется гепатомегалией и задержкой психического развития.
Анри́-Жери́ Э́рс — бельгийский физиолог и биохимик, профессор Лувенского католического университета.

Болезнь Таруи, также гликогеноз VII типа — наследственное метаболическое заболевание типа гликогенозов, выражающееся в дефиците фермента фосфофруктокиназы в мышечной ткани. Болезнь названа в честь японского врача Сэйитиро Таруи, который впервые описал заболевание в 1965 году.
Болезнь Хага — гликогеноз 9 типа. Данное заболевание относится к группе гликогеновых болезней. Причиной является мутация гена PHKA2, отвечающего за синтез α2 -субъединицы фермента киназы фосфорилазы. Этот ген располагается на коротком плече 10-й хромосомы в области Хр22.13. Данное заболевание относится к печеночной форме гликогенозов, то есть происходит нарушение использования гликогена, который необходим для поддержания нормального уровня глюкозы в крови. Болезнь описана врачом Г. Хагом.