
Лизосо́ма — окружённая мембраной клеточная органелла, в полости которой поддерживается кислая среда и находится множество растворимых гидролитических ферментов. Лизосома отвечает за внутриклеточное переваривание макромолекул, в том числе при аутофагии; лизосома способна к секреции своего содержимого во время экзоцитоза; также лизосома участвует в некоторых внутриклеточных сигнальных путях, связанных с метаболизмом и ростом клетки.
Болезнь Краббе — редкое наследственное заболевание, при котором поражается миелиновая оболочка нервных волокон. Наследуется аутосомно-рецессивно. Болезнь названа в честь датского невролога Краббе, который описал её в 1916 году.

Болезнь Баттена — редкое смертельное нейродегенеративное рецессивно наследуемое заболевание, начинающееся в детстве, наиболее частое из группы восковидных липофусцинозов нейронов (ВЛН), относящихся к лизосомным болезням накопления. Хотя обычно это название употребляется в отношении заболевания с детства, некоторые учёные называют так все ВЛН. Исторически восковидные липофусцинозы нейронов делятся на детские, позднедетские, юношеские и взрослые. В ходе заболевания в клетках нервной системы скапливаются жировые вещества, что приводит к потере речи, нарушениям зрения, слабоумию, эпилепсии.

Лизосо́мные боле́зни накопле́ния — общее название группы весьма редких наследственных заболеваний, вызванных нарушением функции внутриклеточных органелл лизосом. Эти одномембранные органоиды являются частью эндомембранной системы клетки и специализируются на внутриклеточном расщеплении веществ: гликогена, гликозаминогликанов, гликопротеинов и других. Лизосомные болезни накопления вызываются генетически обусловленным дефицитом ферментов лизосом, что приводит к накоплению макромолекул, являющихся субстратом этих ферментов, в различных органах и тканях организма.

Сфинголипидо́зы группа лизосомных болезней накопления, связанных с нарушением метаболизма сфинголипидов, относится к классу болезней накопления липидов (липидозов). Основными представителями этой группы являются болезнь Ниманна — Пика, болезнь Фабри, болезнь Краббе, болезнь Гоше, болезнь Тея — Сакса и метахроматическая лейкодистрофия. Они, как правило, наследуется по аутосомно-рецессивному типу, однако в частности, болезнь Фабри — редкое генетически детерминированное заболевание с Х-сцепленным рецессивным типом наследования.
Синдро́м Гу́рлер — тяжёлое наследственное заболевание из группы мукополисахаридозов, относящихся к лизосомным болезням накопления. Характеризуется недостаточностью альфа-L-идуронидазы — фермента лизосом, участвующего в катаболизме кислых мукополисахаридов, которые составляют основу межклеточного вещества соединительной ткани.

Воскови́дные липофусцино́зы нейро́нов — общее название широкой группы нейродегенеративных наследственных заболеваний, относящихся к лизосомным болезням накопления. Симптомы болезней данной группы обусловлены чрезмерным накоплением пигмента липофусцина в лизосомах нервных клеток и многих других тканей организма, включая печень, селезёнку, миокард, почки. Избыточное отложение липофусцина в лизосомах вызывает зеленовато-жёлтое окрашивание при микроскопии в ультрафиолетовых лучах.
Боле́знь Бильшо́вского — Янско́го — поздняя инфантильная (детская) форма восковидного липофусциноза нейронов, которая развивается на фоне дефицита лизосомного фермента трипептидил-пептидазы-1. Относится к группе лизосомных болезней накопления.
Синдро́м Шейе — тяжёлое наследственное заболевание из группы мукополисахаридозов, относящихся к лизосомным болезням накопления. Характеризуется недостаточностью альфа-L-идуронидазы — фермента лизосом, участвующего в катаболизме кислых мукополисахаридов, которые составляют основу межклеточного вещества соединительной ткани. Заболевание редкое, проявляется в детском возрасте.
Синдром Марото — Лами — редкая наследственная болезнь, одна из форм мукополисахаридоза из группы лизосомных болезней накопления, биохимически связанная с дефицитом фермента лизосом N-Ацетилгексозамин-4-сульфатсульфатазы.

GM2-ганглиозидо́з — тяжёлое наследственное заболевание, развивающееся в результате дефицита или недостаточной активности фермента гексозаминидазы и накопления в клетках ганглиозидов. Относится к лизосомным болезням накопления и имеет три варианта, связанные с мутациями в разных генах, которые оказывают вляние на активность общей гексозаминидазы. Два варианта заболевания больше известны под индивидуальными именами, полученными в честь авторов, впервые описавших их клиническую картину: болезнь Тея — Сакса, болезнь Сандхоффа. Третий вариант этой болезни носит название GM2 ганглиозидоз, вариант АБ. Болезнь Тея — Сакса вызвана мутацией в гене HEXA, кодирующий альфа-субъединицу гексоаминидазы А. Болезнь Сандхоффа вызвана мутацией в гене HEXB, кодирующий бета-субъединицу гексоаминидаз А и Б. GM2 ганглиозидоз, АБ вариант связан с нарушением в гене GM2, кодирующий белок-активатор GM2A.
GM2 ганглиозидо́з, вариа́нт АБ (англ. GM2-gangliosidosis, AB variant) — редкое аутосомно-рецессивное нарушение обмена веществ, связанное с мутацией гена GM2A. Характеризуется нормальной активностью β-гексозаминидаз А и Б и обусловлено недостаточностью активатора (белкового кофактора), необходимого для реализации ферментной активности в отношении субстрата. Заболевание клинически проявляется прогрессирующим разрушением нервных клеток головного и спинного мозга.

Синдром Сандхоффа — наследственное заболевание, вариант 0 GM2-ганглиозидоза из группы лизосомных болезней накопления — нарушение липидного обмена, вызванное наследственным дефицитом (недостаточностью) ферментов бета-гексозаминидазы А и Б.
Синдро́м Сла́я — редкое наследственное заболевание из группы мукополисахаридозов, относящееся к лизосомным болезням накопления, характеризуется дефицитом фермента лизосом β-глюкуронидазы. В свою очередь, дефицит β-глюкуронидазы ведёт к накоплению определённых сложных углеводов (мукополисахаридов) во многих тканях и органах тела человека.

I-кле́точная боле́знь — наследственное заболевание из группы муколипидозов, относящееся к лизосомным болезням накопления. Клиническая картина развивается в результате дефекта фосфотрансферазы. Метаболическая роль этого фермента, принимающего участие в посттрансляционном синтезе олигосахаридной части лизосомных ферментов, заключается в синтезе специфичной метки катаболических ферментов лизосом, расщепляющих олигосахариды, липиды и гликозаминогликаны внутри клетки.
Мно́жественная сульфата́зная недоста́точность — очень редкое аутосомно-рецессивное наследственное заболевание из группы лизосомных болезней накопления. Болезнь развивается в результате недостаточности формилглицинсинтезирующего фермента, активирующего некоторые сульфатазы, среди которых три изофермента арилсульфатазы A, B, C и другие сульфатазы. Клиническая картина заболевания напоминает позднюю инфантильную форму метахроматической лейкодистрофии и мукополисахаридоза.
Сиалолипидо́з — аутосомно-рецессивное наследственное заболевание из группы муколипидозов, относящееся к лизосомным болезням накопления. Клиническая картина отличается разнообразной симптоматикой, включающей задержку психомоторного развития и различные глазные аберрации. Заболевание обусловлено различными мутациями гена MCOLN1, расположенного на длинном плече 19-й хромосомы (19q13.3-p13.2), который кодирует неселективный катионный канал муколипина-1. Результатом данной генной мутации является нарушение клеточных функций, которое ведёт к патологии нервной системы посредством неизвестного механизма.

Маннозидо́зы — редкие наследственные заболевания из группы лизосомных болезней накопления с аутосомно-рецессивным типом наследования.
Боле́знь Ку́фса — редкое наследственное нейродегенеративное заболевание с летальным исходом из группы лизосомных болезней накопления, которое развивается на фоне дефицита фермента лизосом и наследуется по аутосомно-рецессивному типу.
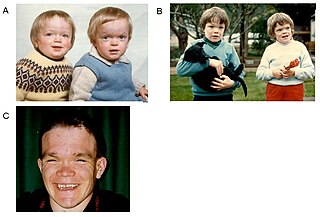
Альфа-маннозидо́з — редкое аутосомно-рецессивное наследственное заболевание из группы лизосомных болезней накопления, связанное с нарушением метаболизма олигосахаридов в результате снижения активности фермента лизосом альфа-маннозидазы. Также заболевание наносит ущерб в животноводстве.
