
Инфа́ркт миока́рда возникает из-за полной или частичной окклюзии артерии, питающей сердце. Нарушение поступления крови к сердцу может привести к серьёзному повреждению или гибели сердечной мышцы. Инфаркт миокарда — состояние, угрожающее жизни, поэтому важно вызвать скорую помощь как можно скорее, если вы подозреваете его у себя или другого человека.
Психо́з, или психоти́ческое расстро́йство — явно выраженное нарушение психической деятельности, при котором психические реакции совершенно не соответствуют происходящему, что отражается в расстройстве восприятия действительности и дезорганизации поведения.
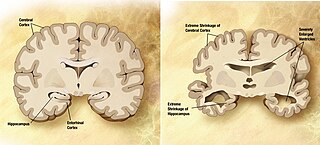
Боле́знь Альцгеймера — наиболее распространённая форма деменции, нейродегенеративное заболевание, впервые описанное в 1907 году немецким психиатром Алоисом Альцгеймером (1864—1915). Как правило, она обнаруживается у людей старше 65 лет, но существует и ранняя болезнь Альцгеймера — редкая форма заболевания. Общемировая заболеваемость на 2006 год оценивалась в 26,6 млн человек, а к 2050 году число больных может вырасти вчетверо.
Болезнь Ка́наван, также известная как болезнь Канаван — ван Богерта, спонгиозная младенческая дегенерация, спонгиозная дегенерация белого вещества мозга, болезнь Кэнэвэн — ван-Богарта — Бертрана, дегенерация ЦНС губчатая, — наследственное заболевание, характеризующееся прогрессирующим поражением нервных клеток мозга. Принадлежит к группе генетически обусловленных лейкодистрофий. При лейкодистрофии происходит разрушение миелиновой оболочки нервных волокон. Болезнь наиболее рапространена среди евреев-ашкеназов.

Анандамид — органическое соединение, эндогенный каннабиноидный нейротрансмиттер. Это соединение содержится во многих органах животных и человека. Соединение было впервые выделено в 1992 году на кафедре химии природных веществ Еврейского университета в Иерусалиме группой исследователей под руководством профессора Рафаэля Мешулама и сотрудниками его лаборатории Вильямом Девейном и Люмиром Ханушем ; в дальнейшем в этой же лаборатории же была определена его структура. Название вещества, предложенное авторами этой статьи, было взято из санскрита — ананда переводится как «блаженство» или «идеальное счастье», а слово амид обозначает химический класс вещества.
Боково́й (латера́льный) амиотрофи́ческий склеро́з (БАС; также известен как боле́знь мото́рных нейро́нов, мотонейро́нная боле́знь, боле́знь Шарко́, в англоязычных странах — болезнь Лу Ге́рига — прогрессирующее, неизлечимое дегенеративное заболевание центральной нервной системы, при котором происходит поражение как верхних, так и нижних двигательных нейронов, что приводит к параличам и последующей атрофии мышц.

Макулодистрофи́я — общее название для группы заболеваний, при которых поражается сетчатка глаза и нарушается центральное зрение. В основе макулодистрофии лежит патология сосудов и ишемия центральной зоны сетчатки, которая отвечает за центральное зрение. Возрастная макулодистрофия — одна из самых частых причин слепоты у людей старше 55 лет.
Болезнь Вильсона — Коновалова — врождённое или приобретенное нарушение метаболизма меди, приводящее к тяжелейшим поражениям центральной нервной системы и внутренних органов. Диагностируется у 5-10 % больных циррозом печени дошкольного и школьного возраста. Заболевание передаётся по аутосомно-рецессивному типу. Ген ATP7B, мутации которого вызывают заболевание, расположен на 13-й хромосоме.

Синдром Дента — редкий Х-сцепленный рецессивный гипофосфатемический рахит, который поражает проксимальные почечные канальцы почек. Вызван мутацией гена хлоридного канала (CLCH5-ген), локализованного на хромосоме Хр11.22. Проявляется синдромом Фанкони, тубулярной протеинурией, нефрокальцинозом, нефролитиазом и прогрессированием до хронической почечной недостаточности. Лабораторно отмечают повышение кальцитриола, гиперкальциурия, гипофосфатемия.
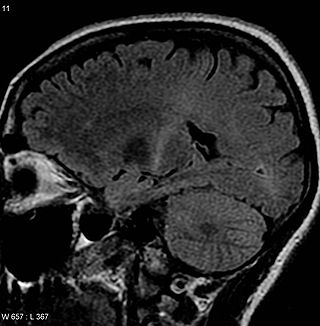
Опухоли ЦНС — новообразования центральной нервной системы. Образуются в результате неконтролируемого деления клеток, в большинстве случаев — клеток глии, поскольку собственно зрелые нейроны теряют способность к делению.
Ацерулоплазминемия — редкое заболевание, вызванное мутациями гена, кодирующего синтез церулоплазмина. Данный белок, основная функция которого заключается в транспорте меди, также участвует в метаболизме железа, и мутации, нарушающие эту функцию церулоплазмина, приводят к токсичным отложениям железа в мозге, печени, поджелудочной железе.

Синдром MELAS — прогрессирующее нейродегенеративное заболевание, характеризующееся проявлениями, перечисленными в названии, и сопровождается полиморфной симптоматикой — диабетом, судорогами, снижением слуха, сердечными заболеваниями, низким ростом, эндокринопатиями, непереносимостью физических нагрузок и нейропсихиатрическими отклонениями. В каждом конкретном случае набор симптомов и их тяжесть может сильно отличаться, поскольку синдром связан с мутациями во многих генах: MTTL1, MTTQ, MTTH, MTTK, MTTS1, MTND1, MTND5, MTND6, MTTS2. Мутации могут возникать впервые у конкретного пациента, либо наследоваться по материнской линии. Всего к 2009 году было обнаружено 23 миссенсных точечных мутаций и 4 делеции мтДНК, приводящих к MELAS, однако продолжаются сообщения о новых пациентах с симптомами расстройства при отсутствии известных мутаций.
Аутоантитела — антитела, способные взаимодействовать с аутоантигенами, то есть с антигенами собственного организма. Могут образовываться спонтанно или вследствие перенесенных инфекций.

Неалкогольная жировая болезнь печени — частный случай стеатогепатоза, возникающего у людей, не злоупотребляющих алкоголем. Он связан с инсулинорезистентностью и метаболическим синдромом. Лечение включает в себя снижение веса, приём сахароснижающих препаратов. Неалкогольный стеатогепатит — наиболее тяжёлая форма НЖБП, приводящая к циррозу печени.

Воскови́дные липофусцино́зы нейро́нов — общее название широкой группы нейродегенеративных наследственных заболеваний, относящихся к лизосомным болезням накопления. Симптомы болезней данной группы обусловлены чрезмерным накоплением пигмента липофусцина в лизосомах нервных клеток и многих других тканей организма, включая печень, селезёнку, миокард, почки. Избыточное отложение липофусцина в лизосомах вызывает зеленовато-жёлтое окрашивание при микроскопии в ультрафиолетовых лучах.
Болезнь Штаргардта или жёлтопятнистая абиотрофия сетчатки , является унаследованной формой ювенильной макулярной дегенерации, которая вызывает прогрессирующее ухудшение зрения, как правило, до точки официальной слепоты. Прогрессирование обычно начинается в возрасте от шести до двенадцати лет, и выравнивается вскоре после быстрого снижения остроты зрения. Несколько генов ассоциированы с этим расстройством. Симптомы обычно развиваются до возраста двадцати лет, и включают волнистое видение, слепые пятна, размытость, нарушение цветового зрения, и трудности с адаптацией при тусклом освещении.

ATP8B1 — фермент, продукт гена человека ATP8B1. Белок связан с развитием таких заболеваний, как прогрессирующий наследственный внутрипечёночный холестаз 1-го типа и доброкачественный рецидивирующий внутрипечёночный холестаз.

Мутация H63D вызывает в 63-й позиции замену аминокислоты гистидин на аспартат и приводит к нарушению пространственной структуры белка HFE. Такой белок не способен связываться с рецептором трансферрина 1. Гетерозиготных носителей мутации около 15 % в популяциях русских и среднеевропейцев. Гомозиготные мутации H63D гена HFE иногда являются причиной гемохроматоза, однако они связаны с возникновением других состояний, таких как гипотрансферринемия, дисфункция печени, проблемы с костями и суставами, сахарный диабет, болезни сердца, дисбаланс гормонов, поздняя порфирия, бесплодие, инсульт, нейродегенеративные заболевания и повреждения мозга, некоторые виды рака, заболевания вен и периферических артерий.
Синдром Экарди — Гутьер — является редким, обычно начинающимся в раннем детстве, воспалительным заболеванием, чаще всего поражающим мозг и кожу. У большинства пациентов наблюдаются серьёзные соматические проявления и умственная отсталость. Клинические признаки синдрома могут имитировать признаки внутриутробной инфекции, а некоторые симптомы также совпадают с системной красной волчанкой. Первое описание восьми случаев синдрома было сделано в 1984 г., болезнь получила название «синдром Экарди-Гутьер» в 1992 году, первая конференция, посвященная этому заболеванию, была проведена в Павии в 2001 году.
Ранняя болезнь Альцгеймера, также болезнь Альцгеймера с ранним началом — болезнь Альцгеймера, диагностируемая в возрасте до 65 лет. Это необычная форма болезни, на которую приходится всего 5—10 % всех случаев заболеваний. Около 60 % имеют положительный семейный анамнез болезни Альцгеймера, и 13 % из них наследуются по аутосомно-доминантному типу. Большинство случаев болезни Альцгеймера с ранним началом имеют те же черты, что и обычная форма, и не вызваны известными генетическими мутациями.
