
Насле́дственные заболева́ния — заболевания, возникновение и развитие которых связано с различными дефектами и нарушениями в наследственном аппарате клеток. В основе наследственных заболеваний лежат мутации: хромосомные, генные и митохондриальные. Наследственные заболевания могут быть обусловлены мутациями, передаваемыми в семьях по наследству, или мутациями, вновь возникшими в клетках зародышевой линии, в зиготе или на очень ранних этапах развития. Наследственные болезни многочисленны и разнообразны по проявлениям.

Альбинизм — наследственное заболевание, полное или почти полное отсутствие пигмента меланина или хлорофилла. Проявляется отсутствием нормальной вида окраски кожи, волос, шерсти, радужной и пигментной оболочек глаз, зелёных частей растений.

Болезнь Гентингтона — аутосомно-доминантное генетическое заболевание нервной системы, характеризующееся постепенным началом обычно в возрасте 30—50 лет и сочетанием прогрессирующего хореического гиперкинеза и психических расстройств. Заболевание вызывается умножением кодона CAG в гене HTT. Этот ген кодирует 350-kDa белок хантингтин с неизвестной функцией. В гене дикого типа у разных людей присутствует разное количество CAG-повторов, однако, когда число повторов превышает 36, развивается болезнь. Нейроморфологическая картина характеризуется атрофией стриатумa, а на поздней стадии также атрофией коры головного мозга.
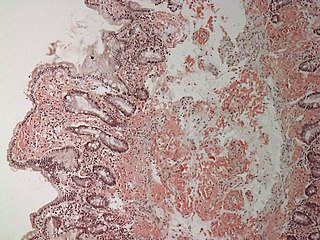
Амилоидóз — нарушение белкового обмена, сопровождающееся образованием и отложением в тканях специфического белково-полисахаридного комплекса — амилоида.

Митохондриа́льные заболева́ния — группа наследственных заболеваний, связанных с дефектами в функционировании митохондрий, приводящими к нарушениям энергетических функций в клетках эукариот, в частности, человека. На 2021 год известно более 350 генов, приводящих к таким заболеваниям.

Боле́знь Тея — Са́кса (GM2 ганглиозидоз, ранняя детская амавротическая идиотия) — редкое наследственное заболевание с аутосомно-рецессивным типом наследования, поражающее центральную нервную систему (спинной и головной мозг, а также менингеальные оболочки). Относится к группе лизосомных болезней накопления. Названо в честь британского офтальмолога Уоррена Тея (англ. Warren Tay, 1843—1928) и американского невролога Бернарда Сакса (англ. Bernard Sachs, 1858—1944), которые впервые описали это заболевание независимо друг от друга в 1881 и 1887 годах соответственно.

Синдро́м Ма́ртина — Белл — наследственное заболевание.
Синдро́м (болезнь) де То́ни — Дебре́ — Фанко́ни — врождённое заболевание, наследуется по аутосомно-рецессивному типу. Комплекс биохимических и клинических проявлений поражения проксимальных почечных канальцев с нарушением канальцевой реабсорбции фосфата, глюкозы, аминокислот и бикарбоната. Одно из рахитоподобных заболеваний.
Атаксия Фридрейха — аутосомно-рецессивное заболевание, характеризующееся дегенеративным повреждением нервной системы вследствие наследуемой мутации в гене FXN, кодирующем белок фратаксин. Заболевание наследуется по аутосомно-рецессивному типу. Заболевание названо в честь немецкого врача Николауса Фридрейха, который первым описал её в 1860 году.

Микросателли́ты, или короткие тандемные (простые) повторы, — варьирующие участки (локусы) в ядерной ДНК и ДНК органелл, состоящие из тандемно повторяющихся мономеров длиной меньше 9 пар оснований и образующие поля менее 1 тысячи пар оснований. Являются широко распространёнными молекулярными маркерами в генетических и геномных исследованиях.
Втори́чные фо́рмы са́харного диабе́та — разнородная группа заболеваний, к которой относится сахарный диабет, встречающийся на фоне другой клинической патологии, которая может и не сочетаться с сахарным диабетом. Для большинства заболеваний из этой группы этиологические факторы раскрыты. Кроме того, к этой группе заболеваний также относят и некоторые генетические (наследственные) синдромы, в том числе аномалии инсулиновых рецепторов. При вторичных формах сахарного диабета отсутствуют ассоциации с HLA-антигенами, данные за аутоиммунное поражение и антитела к островковой ткани поджелудочной железы.
Нейродегенеративные заболевания — группа в основном медленно прогрессирующих, наследственных или приобретённых заболеваний нервной системы. Общим для этих заболеваний является прогрессирующая гибель нервных клеток (нейродегенерация), ведущая к различным неврологическим симптомам — прежде всего, к деменции и нарушению движений. Заболевания могут наступить в различном возрасте, протекают диффузно или генерализированно, гистологически определяется специфический тип изменений.

Гаргоили́зм — характерная для болезни Пфаундлера — Гурлер и некоторых других типов мукополисахаридоза внешность: грубые черты лица: широкая и приплюснутая переносица, широко расставленные глаза, широкий с большими ноздрями нос, толстые губы и язык, открытый рот с маленькими редкими зубами; форма черепа нередко напоминает киль лодки. Заболевание обусловлено наследственной патологией соединительной ткани, проявляющейся поражением костей, суставов, глаз, внутренних органов и нервной системы. Понятие гаргоилизм берёт начало от фр. gargouille (гаргу́лья), обозначающего раструбы старинных водосточных труб средневековых соборов, украшавшиеся рельефными изображениями фантастических фигур с отталкивающим, причудливым лицом (химер).
GM2-ганглиозидо́з — тяжёлое наследственное заболевание, развивающееся в результате дефицита или недостаточной активности фермента гексозаминидазы и накопления в клетках ганглиозидов. Относится к лизосомным болезням накопления и имеет три варианта, связанные с мутациями в разных генах, которые оказывают вляние на активность общей гексозаминидазы. Два варианта заболевания больше известны под индивидуальными именами, полученными в честь авторов, впервые описавших их клиническую картину: болезнь Тея — Сакса, болезнь Сандхоффа. Третий вариант этой болезни носит название GM2 ганглиозидоз, вариант АБ. Болезнь Тея — Сакса вызвана мутацией в гене HEXA, кодирующий альфа-субъединицу гексоаминидазы А. Болезнь Сандхоффа вызвана мутацией в гене HEXB, кодирующий бета-субъединицу гексоаминидаз А и Б. GM2 ганглиозидоз, АБ вариант связан с нарушением в гене GM2, кодирующий белок-активатор GM2A.

Синдро́м ван Богарта — Шерера — Эпште́йна — редкое аутосомно-рецессивное наследственное заболевание, проявляющееся расстройством липидного обмена, которое приводит к накоплению липидов в клетках. Причиной заболевания является мутация в гене CYP27A1, кодирующего митохондриальный фермент стерол-27-гидроксилазу.

Аутосо́мно-рецесси́вное насле́дование — свойственный диплоидным эукариотам тип наследования признака, контролируемого рецессивными аллелями аутосомного гена. Для проявления мутации или болезни с таким типом наследования мутантный аллель, локализованный в аутосоме, должен быть унаследован от обоих родителей. Иными словами, мутация проявляется только в гомозиготном состоянии, то есть тогда, когда обе копии гена, расположенные на гомологичных аутосомах, являются повреждёнными. Если мутация находится в гетерозиготном состоянии, и мутантному аллелю сопутствует нормальный функциональный аллель, то аутосомно-рецессивная мутация не проявляется.

Фукозидо́з — редкое наследственное заболевание из группы лизосомных болезней накопления, связанное с недостаточностью гидролаз, расщепляющих полисахаридные связи. В результате внутри клеток накапливаются два класса макромолекул: гликолипиды и гликопротеины. Клиническая картина связана с мутацией, приводящей к повреждению или дефициту фермента лизосом альфа-L-фукозидазы и характеризуется разнообразными соматическими проявлениями и нарушением функции нервной системы.
Синдро́м Ке́рнса — Се́йра (англ. Kearns–Sayre syndrome, сокращённо KSS) — митохондриальная миопатия с типичным началом до 20-летнего возраста. KSS является более серьёзным синдромным вариантом хронической прогрессирующей внешней офтальмоплегии, синдром, который характеризуется изолированным поражением мышц, контролирующих движения век и контролирующих движения глаз. Это приводит к птозу и офтальмоплегии соответственно. KSS включает в себя триаду уже описанных CPEO, двустороннюю пигментную ретинопатию и блокаду сердца. Другие области участия могут включать в себя мозжечковую атаксию, проксимальную мышечную слабость, глухоту, сахарный диабет, дефицит гормона роста, гипопаратиреоз или другие эндокринные нарушения. В обоих этих заболеваниях вовлечение мышц может начаться односторонним, но всегда развивается в двусторонний дефицит, который является прогрессирующим.
Эта статья относится к регионам в последовательности эукариотической хромосомы. Некоторые регионы могут быть включены более чем в одну группу, так что представленную классификацию не стоит рассматривать как полностью раздельную.