TGFBI — белок человека. Экспрессия TGFBI индуцируется фактором роста TGF-beta. Альтернативное название кератоэпителин свидетельствует об узко выраженной картине экспрессии белка в тканях глаза: он производится клетками эпителия роговицы и стромальными кератоцитами. Ген был впервые описан в 1997 году, изначально получив название BIGH3.

Эпителий роговицы — наружный слой роговой оболочки глаза. У человека эпителий расположен над слоем Боумена, у ряда других млекопитающих — непосредственно над стромой роговицы. Эпителий состоит из нескольких слоёв эпителиальных клеток: у человека в центральной зоне насчитывают пять слоёв, на периферии — до 10. Эпителий роговицы уникален своей прозрачностью и отсутствием кровеносных сосудов; на периферии он сменяется лимбом роговицы, за которым следует конъюнктива.

Эндотелиальная дистрофия роговицы — дистрофия роговой оболочки глаза, при которой отмирают клетки её низшего слоя — заднего эпителия (эндотелия), обеспечивающие откачивание лишней жидкости из стромы. Заболевание встречается чуть чаще у женщин и обычно проявляет себя, когда пациент достигает возраста 30 или 40 лет.
Эпителиальная дистрофия роговицы Лиша - редкая форма дистрофии роговой оболочки человеческого глаза. Впервые описана в 1992 году авторами Lisch et al. В одном исследовании отмечена ассоциация заболевания с хромосомной областью Xp22.3, но пока что не выявлено конкретных генов-кандидатов.
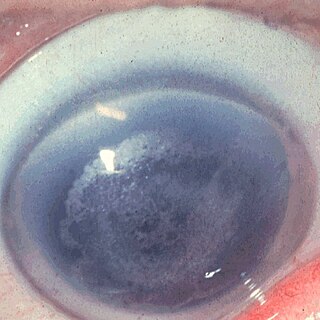
Кристаллическая дистрофия роговицы Шнайдера — редкая форма дистрофии роговой оболочки глаза. Болезнь вызывается мутациями гена UBIAD1. Первые описания кристаллической дистрофии датируются 1950-ми годами.

Врождённая эндотелиальная дистрофия роговицы, тип 2 — редкая форма дистрофии роговой оболочки человека. Наследуется по аутосомно-рецессивному пути, в отличие от первого типа эндотелиальной врождённой дистрофии. Выявлена ассоциация с геном SLC4A11, причём с этим геном также ассоциирован редкий синдром, характеризующийся аутосомно-рецессивной дистрофией роговицы и перцептивной глухотой. Ген SLC4A11 кодирует транспортер, предположительно регулирующий внутриклеточную концентрацию бора.

Боле́знь По́мпе — редкое, наследственное заболевание с аутосомно-рецессивным механизмом, наследования,, связанное с повреждением мышечных, и нервных клеток по всему организму. Клиническая картина данной патологии, обусловлена накоплением гликогена, в лизосомах, вызванным недостаточностью лизосомного фермента — кислой α-1,4-глюкозидазы. Различают быстро прогрессирующую (классическую) и медленно прогрессирующую формы болезни Помпе. Несмотря на широкий спектр клинических проявлений, гликогеноза II типа, в основе всех форм болезни лежит, дефицит одного фермента, кодируемого геном GAA.

GM2-ганглиозидо́з — тяжёлое наследственное заболевание, развивающееся в результате дефицита или недостаточной активности фермента гексозаминидазы и накопления в клетках ганглиозидов. Относится к лизосомным болезням накопления и имеет три варианта, связанные с мутациями в разных генах, которые оказывают вляние на активность общей гексозаминидазы. Два варианта заболевания больше известны под индивидуальными именами, полученными в честь авторов, впервые описавших их клиническую картину: болезнь Тея — Сакса, болезнь Сандхоффа. Третий вариант этой болезни носит название GM2 ганглиозидоз, вариант АБ. Болезнь Тея — Сакса вызвана мутацией в гене HEXA, кодирующий альфа-субъединицу гексоаминидазы А. Болезнь Сандхоффа вызвана мутацией в гене HEXB, кодирующий бета-субъединицу гексоаминидаз А и Б. GM2 ганглиозидоз, АБ вариант связан с нарушением в гене GM2, кодирующий белок-активатор GM2A.
GM2 ганглиозидо́з, вариа́нт АБ (англ. GM2-gangliosidosis, AB variant) — редкое аутосомно-рецессивное нарушение обмена веществ, связанное с мутацией гена GM2A. Характеризуется нормальной активностью β-гексозаминидаз А и Б и обусловлено недостаточностью активатора (белкового кофактора), необходимого для реализации ферментной активности в отношении субстрата. Заболевание клинически проявляется прогрессирующим разрушением нервных клеток головного и спинного мозга.
Вителиформная макулярная дистрофия или вителиформная дистрофия — генетическое расстройство глаз, порождающее прогрессирующую потерю зрения, влияющее на макулярную область сетчатки, которая обеспечивает резкое центральное зрение, необходимое для получения мелких деталей изображения для чтения, вождения автомобиля, и распознавания лиц.

Пигментный ретинит (RP) — наследственное, дегенеративное заболевание глаз, которое вызывает сильное ухудшение зрения и часто слепоту. Прогрессирование RP не является последовательным. Некоторые люди могут иметь симптомы с детства, другие могут заметить симптомы позднее. В общем, чем позже начало, тем быстрее ухудшение зрения. Те, кто не имеют RP имеют 90-градусное периферийное зрение, в то время как люди, имеющие RP имеют периферийное зрение менее 90 градусов.

Периферин 2 представляет собой белок, который у человека кодируется геном PRPH2. Периферин 2 находится в клетках палочек и колбочек сетчатки глаза. Дефекты этого белка являются причиной одной из форм пигментного ретинита, неизлечимой слепоты.

Маннозидо́зы — редкие наследственные заболевания из группы лизосомных болезней накопления с аутосомно-рецессивным типом наследования.
Альфа-маннозидо́з — редкое аутосомно-рецессивное наследственное заболевание из группы лизосомных болезней накопления, связанное с нарушением метаболизма олигосахаридов в результате снижения активности фермента лизосом альфа-маннозидазы. Также заболевание наносит ущерб в животноводстве.
Хроническая прогрессирующая внешняя офтальмоплегия (CPEO), также известная как прогрессирующая внешняя офтальмоплегия (ПЭО), является типом расстройства глаз, характеризующимся медленно прогрессирующей невозможностью перемещения глаз и бровей. Это часто единственная особенность этого митохондриального заболевания, в этом случае термин CPEO может быть использован как диагноз. У других людей, страдающих от митохондриальной болезни, CPEO происходит в рамках синдрома с участием более чем одной части тела, например, синдром Кернс-Сейр. Изредка CPEO может быть вызвана условиями, отличными от митохондриальных заболеваний.
Синдро́м Ке́рнса — Се́йра (англ. Kearns–Sayre syndrome, сокращённо KSS) — митохондриальная миопатия с типичным началом до 20-летнего возраста. KSS является более серьёзным синдромным вариантом хронической прогрессирующей внешней офтальмоплегии, синдром, который характеризуется изолированным поражением мышц, контролирующих движения век и контролирующих движения глаз. Это приводит к птозу и офтальмоплегии соответственно. KSS включает в себя триаду уже описанных CPEO, двустороннюю пигментную ретинопатию и блокаду сердца. Другие области участия могут включать в себя мозжечковую атаксию, проксимальную мышечную слабость, глухоту, сахарный диабет, дефицит гормона роста, гипопаратиреоз или другие эндокринные нарушения. В обоих этих заболеваниях вовлечение мышц может начаться односторонним, но всегда развивается в двусторонний дефицит, который является прогрессирующим.

Член 4 подсемейства A (АВС1) ATP-связывающей кассеты , также известный как ABCA4 или ABCR — белок, который в организме человека кодируется геном ABCA4.

Дистрофия колбочек — наследственное глазное расстройство, характеризующееся потерей колбочек фоторецепторов, ответственных как за центральное, так и цветовое зрение.
Синдро́м Ло́уренса-Му́на. Редкое аутосомно-рецессивное генетическое заболевание, характеризующиеся ожирением, умственной отсталостью, атаксией, дистрофией сетчатки, гипопитуитаризмом, большим количеством пальцев рук или ног. Синдром связан с нарушением функции гипоталамических центров.
Прогрессирующая атрофия сетчатки (PRA) — группа генетических заболеваний, встречающихся у определенных пород собак и, реже, у кошек. Подобно пигментному ретиниту у людей, она характеризуется двусторонней дегенерацией сетчатки, что приводит к прогрессирующей потере зрения, ведущей к слепоте. Болезни этой группы почти во всех породах наследуются как аутосомно-рецессивный признак, за исключением сибирского хаски и бульмастифа. На данный момент не существует лечения от этой группы заболеваний.