
Митохондриа́льные заболева́ния — группа наследственных заболеваний, связанных с дефектами в функционировании митохондрий, приводящими к нарушениям энергетических функций в клетках эукариот, в частности, человека. На 2021 год известно более 350 генов, приводящих к таким заболеваниям.

Синдро́м Праде́ра — Ви́лли — редкое наследственное заболевание, причиной которого является отсутствие отцовской копии участка хромосомы 15q11-13. В этом участке хромосомы 15 находятся гены, в регуляции которых задействован геномный импринтинг. Большинство случаев являются спорадическими, для редких описанных семейных случаев характерно неменделевское наследование.
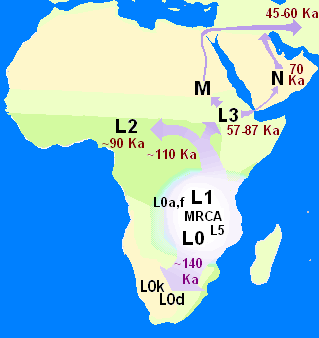
Митохондриальная Ева — имя, данное в популярной культуре женщине, от которой всё современное человечество унаследовало митохондриальную ДНК. Жила в Африке около 200 000 лет назад. Эта женщина стала единственной в своём поколении, чьи потомки по женской линии дожили до наших дней. Параллельно с ней жили и другие женщины, но их митохондриальные ДНК до нашего времени не дошли.

Гаплогруппа J (J-P209) — Y-ДНК-гаплогруппа, распространённая преимущественно на Ближнем Востоке, в Северной Африке, на Кавказе, и на юге Европы, хотя встречается и в иных регионах нашей планеты. В её составе выделяется две крупнейшие ветви :
- Гаплогруппа J1 (Y-ДНК)
- Гаплогруппа J2 (Y-ДНК)
Наследственная оптическая нейропатия LHON Лебера, или атрофия зрительного нерва Лебера , является наследственной митохондриальной дегенерацией ганглионарных клеток (РСК) сетчатки и их аксонов, что приводит к острой или почти острой потере центрального зрения; это влияет преимущественно на молодых мужчин. Тем не менее, LHON передается только по материнской линии, прежде всего, из-за мутаций в митохондриальном геноме, и только яйцеклетка способствует митохондрии в зародыше. LHON, как правило, связана с одной из трех патогенных митохондриальных ДНК (мтДНК) точечных мутаций. Эти мутации действуют на нуклеотиды и репозиционируют 11778 G в А, 3460 G в А и 14484 T в C, соответственно, в ND4, ND1 и Nd6 субъединицах генов в комплексе I окислительного фосфорилирования цепочек в митохондриях. Мужчины не могут передать болезнь своему потомству.

PRP31 — обрабатывающий пре-мРНК фактор 31, гомолог (англ. «PRP31 pre-mRNA processing factor 31 homolog »), также известный как PRPF31 , — белок, кодируемый у человека геном PRPF31.
Доминантная атрофия зрительного нерва , или доминантная атрофия зрительного нерва типа Кьер — аутосомное наследственное заболевание, которое влияет на зрительный нерв, что приводит к снижению остроты зрения и слепоте, начиная с детства. Это обусловлено митохондриальной дисфункцией, опосредующей смерть волокон зрительного нерва. Доминантная атрофия зрительного нерва была впервые описана клинически Баттеном в 1896 году и названа оптической нейропатией Кьера в 1959 году в честь датского офтальмолога Poul Kjer, который исследовал 19 семей с болезнью. Хотя домининантная атрофия зрительного нерва является наиболее распространённой аутосомной наследственной оптической нейропатией и не связана с глаукомой, последняя часто ошибочно диагностируется вместо Кьер.

Ушерин — белки, который у человека кодируется геном USH2A. Этот ген кодирует белок Ушерин, который содержит EGF мотивы ламинина, домен пентаксин, и многие мотивы фибронектина III типа. Данный белок, ассоциированный с базальной мембраной, может играть важную роль в развитии и гомеостазе внутреннего уха и сетчатки. Мутации в пределах этого гена были связаны с синдромом Ушера II-го типа и пигментным ретинитом типа 39. Существуют альтернативные варианты транскриптов сплайсинга, которые кодируют различные изоформы.

Член 4 подсемейства A (АВС1) ATP-связывающей кассеты , также известный как ABCA4 или ABCR — белок, который в организме человека кодируется геном ABCA4.
CHD7 известный также как АТФ-зависимая хеликаза CHD7 — фермент, который у человека кодируется геном CHD7.

Аладин (англ. Aladin), также известный как адракалин — белок, кодируемый у человека геном AAAS (англ. Aladin).
Различные этнолингвистические группы Европы, Кавказа, Центральной Азии, Ближнего Востока, Северной Африки и Южной Азии отличаются распространённостью различных клад гаплогрупп Y-хромосомы. Эти клады обозначаются заглавными латинскими буквами.

Церебральная фолатная недостаточность — синдром, при котором в спинномозговой жидкости пациента снижено содержание 5-метилтетрагидрофолата (5-MTHF), несмотря на нормальное содержание 5-MTHF в сыворотке крови. Набор симптомов варьирует в зависимости от возраста начала заболевания и его причины, и может включать дискинезию, атаксию, эпилептические приступы, задержку психомоторного развития.

Бета-цепь 3 Na+/K+-АТФ-азы (ATP1B3) (англ. Sodium/potassium-transporting ATPase subunit beta-3; CD298) — белок, некаталитический компонент фермента, продукт гена человека ATP1B3. Бета-цепь Na+/K+-АТФ-аза, принадлежит к семейству бета-цепей Na+/K+-АТФ-аз и Н+/К+-АТФаз.
Синдром Смит — Магенис (СМС) — наследственное заболевание, имеет такие проявления, как умственная отсталость, аномалии лица, проблемы со сном и многочисленные поведенческие проблемы, в том числе и такие, как самоповреждение. Частота синдрома Смит — Магенис составляет 1 случай на 15 000-25 000 человек. Синдром Смит — Магенис, как правило, обусловлен микроделецией в коротком плече 17-й хромосомы (17p), и иногда его называют 17p-синдром.

RHCE — мембранный белок, продукт гена RHCE.

Недостаточность 5,10-метенилтетрагидрофолатсинтетазы — редкое расстройство развития нервной системы, вызываемое мутациями гена MTHFS, кодирующего один из ферментов фолатного метаболизма.
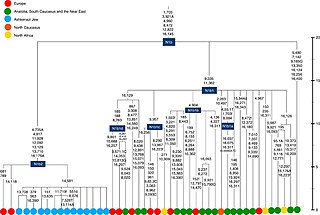
Гаплогруппа N1b — гаплогруппа митохондриальной ДНК человека.

Щелочная фосфатаза, ткань-неспецифический изозим — фермент, который у человека кодируется геном ALPL. Длина полипептидной цепи белка составляет 524 аминокислот, а молекулярная масса — 57 305.
Рибофлавин-зависимая непереносимость физической нагрузки - заболевание, вызываемое мутациями гена SLC25A32, который кодирует митохондриальный транспортер фолата. Назначение рибофлавина значительно улучшает состояние пациентов.