
Фенилкетонури́я — наследственное заболевание группы ферментопатий, связанное с нарушением метаболизма аминокислот, главным образом фенилаланина. Несоблюдение низкобелковой диеты сопровождается накоплением фенилаланина и его токсических продуктов, что приводит к тяжёлому поражению ЦНС, проявляющемуся, в частности, в виде нарушения умственного развития. Одно из немногих наследственных заболеваний, поддающихся успешному лечению.

Синдром Лёша — Нихена — наследственное заболевание, характеризующееся увеличением синтеза мочевой кислоты и вызванное дефектом фермента гипоксантин-гуанинфосфорибозилтрансферазы, который катализирует реутилизацию гуанина и гипоксантина — в результате образуется большее количество ксантина и, следовательно, мочевой кислоты. Частота проявления синдрома 1 к 380000 живорожденных.
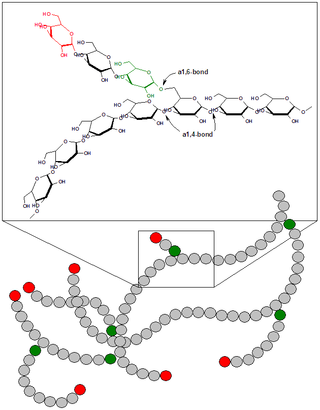
Боле́знь Герса — гепатофосфорилазная недостаточность, гликогеноз, вызванный недостаточностью фосфорилазы печени.
Лейцино́з — врождённое нарушение обмена, ферментопатия.

Синдром Дента — редкий Х-сцепленный рецессивный гипофосфатемический рахит, который поражает проксимальные почечные канальцы почек. Вызван мутацией гена хлоридного канала (CLCH5-ген), локализованного на хромосоме Хр11.22. Проявляется синдромом Фанкони, тубулярной протеинурией, нефрокальцинозом, нефролитиазом и прогрессированием до хронической почечной недостаточности. Лабораторно отмечают повышение кальцитриола, гиперкальциурия, гипофосфатемия.
Рецептор фолиевой кислоты альфа, или рецептор фолиевой кислоты 1, — высокоаффинный клеточный рецептор к фолиевой кислоте и нескольким её производным, относится к семейству белков рецепторов фолиевой кислоты. У человека описано 4 типа рецепторов из этого семейства. В клетке рецептор обеспечивает доставку в клетку 5-метилтетрагидрофолата, кофактора, необходимого для клеточной пролиферации. Вместе с рецептором фолиевой кислоты бета является мишенью для антираковой терапии, так как блокировка транспорта фолата в раковые клетки предотвращает их дальнейшее размножение.

Аргининосукцинат, также аргининоянтарная кислота — непротеиногенная аминокислота, которая синтезируется в цикле мочевины. Является осно́вной аминокислотой.

Врождённый гипотирео́з — гетерогенная по этиологии группа заболеваний щитовидной железы, проявляющихся сразу после рождения и характеризующихся частичным или полным выпадением её функции. Врождённый и приобретённый гипотиреоз в детском возрасте имеет сходную клиническую картину — преобладает торможение всех функций организма: ослаблена деятельность ряда органов и систем, отмечается вялость обменных процессов и наличие трофических расстройств.
Пури́новый обме́н — совокупность протекающих в живых организмах процессов синтеза и распада пуринов и пуриновых нуклеотидов.

Лизосо́мные боле́зни накопле́ния — общее название группы весьма редких наследственных заболеваний, вызванных нарушением функции внутриклеточных органелл лизосом. Эти одномембранные органоиды являются частью эндомембранной системы клетки и специализируются на внутриклеточном расщеплении веществ: гликогена, гликозаминогликанов, гликопротеинов и других. Лизосомные болезни накопления вызываются генетически обусловленным дефицитом ферментов лизосом, что приводит к накоплению макромолекул, являющихся субстратом этих ферментов, в различных органах и тканях организма.

Боле́знь По́мпе — редкое, наследственное заболевание с аутосомно-рецессивным механизмом, наследования,, связанное с повреждением мышечных, и нервных клеток по всему организму. Клиническая картина данной патологии, обусловлена накоплением гликогена, в лизосомах, вызванным недостаточностью лизосомного фермента — кислой α-1,4-глюкозидазы. Различают быстро прогрессирующую (классическую) и медленно прогрессирующую формы болезни Помпе. Несмотря на широкий спектр клинических проявлений, гликогеноза II типа, в основе всех форм болезни лежит, дефицит одного фермента, кодируемого геном GAA.
Недоста́точность ки́слой фосфата́зы — редкое наследственное заболевание из группы лизосомных болезней накопления. Клиническая симптоматика дефицита кислой фосфатазы развивается в раннем возрасте. Характеризуется гепатомегалией и задержкой психического развития.

Бета-маннозидоз — редкое аутосомно-рецессивное наследственное заболевание из группы лизосомных болезней накопления, связанное с нарушением метаболизма олигосахаридов в результате снижения активности фермента лизосом бета-маннозидазы.
Анри́-Жери́ Э́рс — бельгийский физиолог и биохимик, профессор Лувенского католического университета.

Церебральная фолатная недостаточность — синдром, при котором в спинномозговой жидкости пациента снижено содержание 5-метилтетрагидрофолата (5-MTHF), несмотря на нормальное содержание 5-MTHF в сыворотке крови. Набор симптомов варьирует в зависимости от возраста начала заболевания и его причины, и может включать дискинезию, атаксию, эпилептические приступы, задержку психомоторного развития.

Фолиновая кислота (лейковорин) — витамер фолиевой кислоты, применяемый для коррекции токсического действия метотрексата и пириметамина, в терапии колоректального рака, а также для коррекции фолатной недостаточности и при отравлениях метанолом. Принимается перорально либо в виде внутримышечных или внутривенных инъекций.

Недостаточность декарбоксилазы ароматических аминокислот - редкое аутосомно-рецессивное генетическое заболевание, вызываемое мутациями гена DDC, кодирующего соответствующий фермент.
Пиридоксин-зависимая эпилепсия - тяжелая форма эпилепсии, при которой приступы с трудом поддаются контролю обычными противоэпилептическими препаратами. Заболевание развивается из-за мутаций гена ALDH7A1, приводящих к ускоренной деактивации активной формы витамина B6 в организме. Как правило, приступы начинаются вскоре после рождения, реже - через несколько месяцев, крайне редко - через несколько лет. Раннее назначение больших доз витамина B6 позволяет купировать приступы.
Комбинированная малоновая и метилмалоновая ацидурия (КМАММА), также называемая комбинированной малоновой и метилмалоновой ацидемией — это наследственное метаболическое заболевание, характеризующееся повышенным уровнем малоновой кислот и метилмалоновой кислот. Некоторые исследователи предположили, что КМАММА может быть одной из самых распространенных форм метилмалоновой ацидемии и, возможно, одной из самых распространенных врожденных ошибок метаболизма. Из-за того, что диагноз ставится нечасто, чаще всего он остается невыявленным.

Аргининосукцинат-лиаза — фермент, из семейства амидин-лиазы, катализирующий реакцию негидролитического обратимого расщепления молекулы аргининосукцината на аргинин и фумарат. Схема реакции: