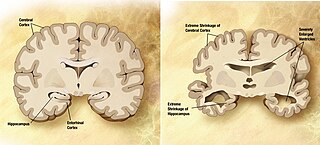
Боле́знь Альцгеймера — наиболее распространённая форма деменции, нейродегенеративное заболевание, впервые описанное в 1907 году немецким психиатром Алоисом Альцгеймером (1864—1915). Как правило, она обнаруживается у людей старше 65 лет, но существует и ранняя болезнь Альцгеймера — редкая форма заболевания. Общемировая заболеваемость на 2006 год оценивалась в 26,6 млн человек, а к 2050 году число больных может вырасти вчетверо.

Спина́льная мы́шечная атрофи́я — разнородная группа наследственных заболеваний, протекающих с поражением / потерей двигательных нейронов передних рогов спинного мозга.

Болезнь Гентингтона — аутосомно-доминантное генетическое заболевание нервной системы, характеризующееся постепенным началом обычно в возрасте 30—50 лет и сочетанием прогрессирующего хореического гиперкинеза и психических расстройств. Заболевание вызывается умножением кодона CAG в гене HTT. Этот ген кодирует 350-kDa белок хантингтин с неизвестной функцией. В гене дикого типа у разных людей присутствует разное количество CAG-повторов, однако, когда число повторов превышает 36, развивается болезнь. Нейроморфологическая картина характеризуется атрофией стриатумa, а на поздней стадии также атрофией коры головного мозга.

Болезнь Гоше́ — наследственное заболевание типа сфинголипидозов, является самой распространённой из лизосомных болезней накопления. Развивается в результате недостаточности фермента глюкоцереброзидазы, которая приводит к накоплению глюкоцереброзида во многих тканях, включая селезёнку, печень, почки, лёгкие, мозг и костный мозг. Заболевание связано с рецессивной мутацией в гене GBA, расположенном в 1-й хромосоме, и поражает как мужчин, так и женщин. Заболевание названо в честь французского врача Филиппа Гоше, который первым описал его в 1882.
Боково́й (латера́льный) амиотрофи́ческий склеро́з (БАС; также известен как боле́знь мото́рных нейро́нов, мотонейро́нная боле́знь, боле́знь Шарко́, в англоязычных странах — болезнь Лу Ге́рига — прогрессирующее, неизлечимое дегенеративное заболевание центральной нервной системы, при котором происходит поражение как верхних, так и нижних двигательных нейронов, что приводит к параличам и последующей атрофии мышц.

Предшественник бета-амилоида — трансмембранный белок, экспрессируемый во многих тканях и концентрирующийся в синапсах нейронов.Его основная функция неизвестна, Хотя он был вовлечен в качестве регулятора образования синапсов, нейропластичности, антимикробной активности и транспорта железа. Наиболее известный тем, что его фрагмент, бета-амилоид, является основным составляющим амилоидных бляшек при болезни Альцгеймера.

Пресенилины — семейство трансмембранных белков, составляющих часть протеазного комплекса γ-секретазы. В геноме позвоночных содержатся два гена, кодирующих пресенилины: PSEN1 кодирует пресенилин 1, а PSEN2 — пресенилин 2. У человека эти гены расположены, соответственно, на 14-й и 1-й хромосомах. Оба гена эволюционно консервативны — отмечены лишь небольшие отличия между пресенилинами крысы и человека. В организме нематоды Caenorhabditis elegans также существуют два белка, напоминающие пресенилины и, судя по всему, выполняющие сходные функции, — sel-12 и hop-1.
Пресенилин-2 — один из двух известных белков-пресенилинов человека. У некоторых больных семейной болезнью Альцгеймера отмечаются мутации PSEN1 и PSEN2. В исследованиях эти мутации усиливают синтез бета-амилоида, основного составляющего амилоидных бляшек, обнаруживаемых в мозге больных при посмертном анализе. Альтернативный сплайсинг порождает несколько форм пресенилина-1, на данный момент описаны две формы белка.
Бапинейзумаб — прототип лекарства для лечения болезни Альцгеймера. Представляет собой моноклональное антитело к бета-амилоидным накоплениям. Возможно терапевтическое действие при глаукоме.

Инозитолтрифосфат (IP3) — это водорастворимый вторичный посредник. IP3 образуется в результате распада мембранных фосфолипидов под действием фермента фосфолипазы С. Инозитолтрифосфат вместе с диацилглицерином принимает участие в передаче сигнала в клетке.

Ге́мохромато́з — наследственное, генетически обусловленное заболевание, проявляется нарушением обмена железа с накоплением его в тканях и органах. Железо поглощается из пищи и чрезмерно накапливается в органах и тканях: печени, поджелудочной железе, миокарде, селезёнке, коже, эндокринных железах и других местах. Избыточное накопление железа в организме может спровоцировать развитие ряда заболеваний: цирроз печени, сердечная недостаточность, сахарный диабет, артрит.

Церебролизин (Cerebrolysin) — препарат из головного мозга свиньи. Позиционируется производителем как ноотропное средство и как препарат с нейротрофической активностью для лечения широкого спектра неврологических расстройств.

Боле́знь Фабри́ — редкое генетически детерминированное заболевание с Х-сцепленным типом наследования, из группы лизосомных болезней накопления. Ранее считалось, что тип наследования болезни Фабри — X-сцепленный рецессивный, однако на современном этапе накоплено достаточно данных, чтобы считать тип наследования болезни Фабри X-сцепленным доминантным с неполной пенетрантностью у женщин. Данное заболевание вызвано нарушением метаболизма сфинголипидов и обладает широким спектром клинических симптомов.

Бета-амило́иды — общее название для нескольких пептидов, состоящих из примерно 40 аминокислотных остатков и образующихся из трансмембранного белка — предшественника бета-амилоида. Основными разновидностями являются пептид из 40 аминокислотных остатков (Aβ40) и пептид из 42 аминокислотных остатков (Aβ42). Их роль в нормальной физиологии остаётся неизвестной, но предполагается, что они могут участвовать в антибактериальной и противогрибковой защите. Пептид Aβ40 не имеет патогенных свойств, но пептид Aβ42 считается одним из основных факторов, провоцирующих болезнь Альцгеймера и часто называется просто бета-амилоидом, без уточнения длины аминокислотной цепочки. В мозге пациента, страдающего болезнью Альцгеймера, этот пептид может образовывать так называемые амилоидные бляшки, состоящие из скоплений пептида, свёрнутого в виде бета-складки. Пептид Aβ42 может также образовывать олигомеры, которые запускают цепные реакции образования амилоидных бляшек и тау-белков по прионному механизму.

Болезнь Шарко — Мари — Тута (ШМТ), или наследственная моторно-сенсорная нейропатия (НМСН) — наследственная периферическая нейропатия с хроническим прогрессирующим течением. При этой болезни больные страдают от слабости и атрофии мышц дистальных отделов конечностей, деформации стоп и кистей, у них наблюдается снижение сухожильных рефлексов, изменение походки, потеря чувствительности в конечностях. В основе клинических проявлений болезни лежит поражение двигательных и чувствительных периферических нервных волокон. Болезнь Шарко — Мари — Тута диагностируется с приблизительной частотой 1 на 2500 человек. Первое проявление болезни чаще всего происходит в подростковом возрасте или в раннем взрослом возрасте. Тяжесть симптомов сильно различается даже среди членов одной семьи с этим заболеванием. Болезнь Шарко — Мари — Тута нередко ведёт к ограничению трудоспособности и к инвалидизации, при этом большинство пациентов имеют нормальную продолжительность жизни. Болезнь Шарко — Мари — Тута является генетически крайне неоднородным заболеванием, симптомы этой болезни могут быть вызваны мутациями в более чем двух десятках генов, хотя большая часть заболеваний вызвана мутациями в генах PMP22, MPZ, GJB1 и MFN2. Наследование болезни чаще всего аутосомно-доминантное, однако может быть аутосомно-рецессивным и Х-сцепленным.

Мутация H63D вызывает в 63-й позиции замену аминокислоты гистидин на аспартат и приводит к нарушению пространственной структуры белка HFE. Такой белок не способен связываться с рецептором трансферрина 1. Гетерозиготных носителей мутации около 15 % в популяциях русских и среднеевропейцев. Гомозиготные мутации H63D гена HFE иногда являются причиной гемохроматоза, однако они связаны с возникновением других состояний, таких как гипотрансферринемия, дисфункция печени, проблемы с костями и суставами, сахарный диабет, болезни сердца, дисбаланс гормонов, поздняя порфирия, бесплодие, инсульт, нейродегенеративные заболевания и повреждения мозга, некоторые виды рака, заболевания вен и периферических артерий.
Гериатри́ческая психиатри́я — отрасль психиатрии, занимающейся изучением, профилактикой и лечением нейродегенеративных, когнитивных нарушений и психических расстройств у людей пожилого возраста. Гериатрическая психиатрия как специальность имеет значительное пересечение со специальностями гериатрической медицины, поведенческой неврологии, нейропсихиатрии, неврологии и общей психиатрии. Гериатрическая психиатрия стала официальной специальностью психиатрии с определённой программой обучения и основными компетенциями.
Колин Луис Мастерс — австралийский невропатолог, изучающий болезнь Альцгеймера и другие нейродегенеративные расстройства. Мастерс — профессор патологии Мельбурнского университета.
Лобно-височная дегенерация — патологический процесс, возникающий при лобно-височной деменции. Характеризуется атрофией лобных и височных долей головного мозга при сохранении теменных и затылочных долей.
Ранняя болезнь Альцгеймера, также болезнь Альцгеймера с ранним началом — болезнь Альцгеймера, диагностируемая в возрасте до 65 лет. Это необычная форма болезни, на которую приходится всего 5—10 % всех случаев заболеваний. Около 60 % имеют положительный семейный анамнез болезни Альцгеймера, и 13 % из них наследуются по аутосомно-доминантному типу. Большинство случаев болезни Альцгеймера с ранним началом имеют те же черты, что и обычная форма, и не вызваны известными генетическими мутациями.