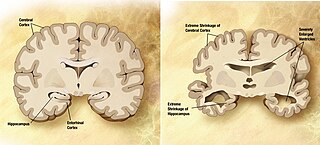
Боле́знь Альцгеймера — наиболее распространённая форма деменции, нейродегенеративное заболевание, впервые описанное в 1907 году немецким психиатром Алоисом Альцгеймером (1864—1915). Как правило, она обнаруживается у людей старше 65 лет, но существует и ранняя болезнь Альцгеймера — редкая форма заболевания. Общемировая заболеваемость на 2006 год оценивалась в 26,6 млн человек, а к 2050 году число больных может вырасти вчетверо.

Болезнь Гентингтона — аутосомно-доминантное генетическое заболевание нервной системы, характеризующееся постепенным началом обычно в возрасте 30—50 лет и сочетанием прогрессирующего хореического гиперкинеза и психических расстройств. Заболевание вызывается умножением кодона CAG в гене HTT. Этот ген кодирует 350-kDa белок хантингтин с неизвестной функцией. В гене дикого типа у разных людей присутствует разное количество CAG-повторов, однако, когда число повторов превышает 36, развивается болезнь. Нейроморфологическая картина характеризуется атрофией стриатумa, а на поздней стадии также атрофией коры головного мозга.
Сплайсинг — процесс вырезания определённых нуклеотидных последовательностей из молекул РНК и соединения последовательностей, сохраняющихся в «зрелой» молекуле, в ходе процессинга РНК. Наиболее часто этот процесс встречается при созревании матричной, или информационной, РНК (мРНК) у эукариот, при этом путём биохимических реакций с участием РНК и белков из мРНК удаляются участки, не кодирующие белок (интроны) и соединяются друг с другом кодирующие аминокислотную последовательность участки — экзоны. Таким образом незрелая пре-мРНК превращается в зрелую мРНК, с которой считываются (транслируются) белки клетки. Большинство генов прокариот, кодирующих белки, не имеют интронов, поэтому у них сплайсинг пре-мРНК встречается редко. У представителей эукариот, бактерий и архей встречается также сплайсинг транспортных РНК (тРНК) и других некодирующих РНК.

Caenorhabditis elegans — свободноживущая почвенная нематода длиной около 1 мм. Исследования этого вида в молекулярной биологии и биологии развития начались в 1974 году работами Сиднея Бреннера. Широко используется как модельный организм в исследованиях по генетике, нейрофизиологии, биологии развития, вычислительной биологии. В 1986 году был полностью описан его коннектом. Геном полностью секвенирован и опубликован в 1998 году. Мартин Чалфи использовал C.elegans при исследовании зелёного флуоресцентного белка.
piРНК — наиболее крупный класс малых некодирующих РНК, экспрессируемых в клетках животных; они обнаружены в комплексах с белками семейства Piwi, за что и получили своё название. piРНК обычно длиннее микроРНК и малых интерферирующих РНК и имеют длину 26—32 нуклеотида, кроме того, в отличие от микроРНК, они не так консервативны. Белки Piwi относятся к большой группе белков Argonaute и экспрессируются почти исключительно в клетках зародышевой линии; они необходимы для поддержания стволовых клеток зародышевой линии, сперматогенеза и репрессии мобильных элементов. Комплексы Piwi с piРНК не только задействованы в сайленсинге ретротранспозонов и других генетических элементов на пост-трансляционном уровне, но имеют и некоторые другие, в значительной мере ещё неописанные эффекты, например, эпигенетические.
Нейрексины — полиморфные мембранные белки, экспрессируемые в нейронах. Открыты в 1992 году. Геномы Drosophila melanogaster и Caenorhabditis elegans содержат по одному гену, кодирующему только альфа-тип белка. У млекопитающих, в том числе мыши и человека, известно три гена нейрексинов: NRXN1, NRXN2, NRXN3, каждый из которых имеет два независимых промотора, в результате чего порождается два типа белков: более крупные альфа-нейрексины, и бета-нейрексины, отличающиеся также конфигурацией внеклеточного участка. Гены нейрексинов человека велики, один только NRXN3 занимает почти 2 % 14й хромосомы. Более того, исключительно эволюционно-консервативный и разнообразный альтернативный сплайсинг порождает множество форм белка: по одной оценке, число их разновидностей в мозге превышает 2000.
Тау-белок принадлежит к группе белков, ассоциированных с микротрубочками (MAP). Альтернативный сплайсинг гена MAPT порождает в организме человека шесть известных изоформ белка. Белок часто встречается в нейронах центральной нервной системы (ЦНС), и редко — в других местах, однако слабо экспрессируется в астроцитах и олигодендроцитах ЦНС.

Мидкин — гепарин-связывающий белок.

Предшественник бета-амилоида — трансмембранный белок, экспрессируемый во многих тканях и концентрирующийся в синапсах нейронов.Его основная функция неизвестна, Хотя он был вовлечен в качестве регулятора образования синапсов, нейропластичности, антимикробной активности и транспорта железа. Наиболее известный тем, что его фрагмент, бета-амилоид, является основным составляющим амилоидных бляшек при болезни Альцгеймера.
Пресенилин-1 - один из двух известных белков-пресенилинов человека. У некоторых больных семейной болезнью Альцгеймера отмечаются мутации PSEN1 и PSEN2. В исследованиях эти мутации усиливают синтез бета-амилоида, основного составляющего амилоидных бляшек, обнаруживаемых в мозге больных при посмертном анализе. Альтернативный сплайсинг порождает несколько форм пресенилина-1.
Пресенилин-2 — один из двух известных белков-пресенилинов человека. У некоторых больных семейной болезнью Альцгеймера отмечаются мутации PSEN1 и PSEN2. В исследованиях эти мутации усиливают синтез бета-амилоида, основного составляющего амилоидных бляшек, обнаруживаемых в мозге больных при посмертном анализе. Альтернативный сплайсинг порождает несколько форм пресенилина-1, на данный момент описаны две формы белка.

Инозитолтрифосфат (IP3) — это водорастворимый вторичный посредник. IP3 образуется в результате распада мембранных фосфолипидов под действием фермента фосфолипазы С. Инозитолтрифосфат вместе с диацилглицерином принимает участие в передаче сигнала в клетке.

PRNP — ген, расположенный на коротком плече 20-й хромосомы. Кодирует нормальный прионный белок (PrPC) и изоформу этого белка — прионный белок PrPSc, связанный с заболеваниями. Пока не ясно, как именно PrPC превращается в PrPSc, как реплицируется PrPSc и почему его накопление вызывает дегенерацию нейронов.
Генами старения можно назвать гены, выключение которых способно замедлить старение. Старение организма является комплексным явлением, в ходе которого происходит ослабление его жизненных функций. Этот процесс неразрывно связан с генетикой организма, и были обнаружены гены, выключение которых приводит к увеличению продолжительности жизни.

SMC5 или белок структурной поддержки хромосом номер пять — это белок, который у человека кодируется геном SMC5.

Холин-О-ацетилтрансфераза, также холин-ацетилтрансфераза, холинацетил-СоА-трансфераза (англ. Choline acetyltransferase, сокр. СhAT, ХАТ, но иногда и CAT) — фермент (КФ 2.3.1.6), из семейства ацилтрансфераз (класс трансферазы), катализирующий реакцию переноса ацетильной группы (CH3-CO) от молекулы ацетил-CoA на молекулу субстрата — холина, с образованием ацетилхолина (АЦХ) и кофермента А, по уравнению:
- ацетил-СоА + холин
 ацетилхолин + CoA-SH.
ацетилхолин + CoA-SH.

Фасцикуляционный и удлиняющий белок дзета-1, сокращённо FEZ-1 или FEZ1, — белок, который у человека кодируется геном FEZ1, локализованным на коротком плече (p-плече) 11-й хромосомы человеческого генома. Длина аминокислотной последовательности этого белка составляет 392 аминокислоты. Молекулярная масса этого белка составляет 45 119 дальтон. Он локализуется в клеточной мембране, цитоплазме, цитоскелете. Относится к фосфопротеинам.

Болезнь Шарко — Мари — Тута (ШМТ), или наследственная моторно-сенсорная нейропатия (НМСН) — наследственная периферическая нейропатия с хроническим прогрессирующим течением. При этой болезни больные страдают от слабости и атрофии мышц дистальных отделов конечностей, деформации стоп и кистей, у них наблюдается снижение сухожильных рефлексов, изменение походки, потеря чувствительности в конечностях. В основе клинических проявлений болезни лежит поражение двигательных и чувствительных периферических нервных волокон. Болезнь Шарко — Мари — Тута диагностируется с приблизительной частотой 1 на 2500 человек. Первое проявление болезни чаще всего происходит в подростковом возрасте или в раннем взрослом возрасте. Тяжесть симптомов сильно различается даже среди членов одной семьи с этим заболеванием. Болезнь Шарко — Мари — Тута нередко ведёт к ограничению трудоспособности и к инвалидизации, при этом большинство пациентов имеют нормальную продолжительность жизни. Болезнь Шарко — Мари — Тута является генетически крайне неоднородным заболеванием, симптомы этой болезни могут быть вызваны мутациями в более чем двух десятках генов, хотя большая часть заболеваний вызвана мутациями в генах PMP22, MPZ, GJB1 и MFN2. Наследование болезни чаще всего аутосомно-доминантное, однако может быть аутосомно-рецессивным и Х-сцепленным.

Сериновая/треониновая протеинкиназа/эндорибонуклеаза Инозитол-требующий фермент 1 α (IRE1α) — это фермент, который у человека кодируется геном ERN1.
Ранняя болезнь Альцгеймера, также болезнь Альцгеймера с ранним началом — болезнь Альцгеймера, диагностируемая в возрасте до 65 лет. Это необычная форма болезни, на которую приходится всего 5—10 % всех случаев заболеваний. Около 60 % имеют положительный семейный анамнез болезни Альцгеймера, и 13 % из них наследуются по аутосомно-доминантному типу. Большинство случаев болезни Альцгеймера с ранним началом имеют те же черты, что и обычная форма, и не вызваны известными генетическими мутациями.
![Solution structure of the human presenilin-1 CTF subunit.[1]](https://upload.wikimedia.org/wikipedia/commons/thumb/2/23/2KR6.pdb.png/260px-2KR6.pdb.png)