
Насле́дственные заболева́ния — заболевания, возникновение и развитие которых связано с различными дефектами и нарушениями в наследственном аппарате клеток. В основе наследственных заболеваний лежат мутации: хромосомные, генные и митохондриальные. Наследственные заболевания могут быть обусловлены мутациями, передаваемыми в семьях по наследству, или мутациями, вновь возникшими в клетках зародышевой линии, в зиготе или на очень ранних этапах развития. Наследственные болезни многочисленны и разнообразны по проявлениям.

Альбинизм — наследственное заболевание, полное или почти полное отсутствие пигмента меланина или хлорофилла. Проявляется отсутствием нормальной вида окраски кожи, волос, шерсти, радужной и пигментной оболочек глаз, зелёных частей растений.

Сетча́тка — внутренняя оболочка глаза, являющаяся периферическим отделом зрительного анализатора; содержит фоторецепторные клетки, обеспечивающие восприятие и преобразование электромагнитного излучения видимой части спектра в нервные импульсы, а также обеспечивает их первичную обработку.

Чиха́нье, чиха́ние, стернута́ция — защитный безусловный рефлекс человека и высших животных, обеспечивающий удаление из верхних дыхательных путей пыли, слизи и других раздражающих агентов путём форсированного выдоха, преимущественно через носоглотку, после короткого глубокого вдоха. В отличие от кашля, при чиханье язык прижимается к мягкому нёбу, поэтому форсированный выдох осуществляется не через рот.

Синдром Ваарденбурга — наследственное заболевание. Имеет следующие клинические признаки: телекант, гетерохромия радужки, седая прядь надо лбом и врождённая тугоухость различной степени.
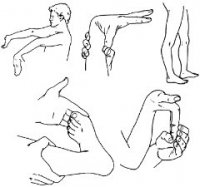
Синдром Э́лерса — Данло́са — это группа наследственных системных дисфункций соединительной ткани, вызванных дефектом в синтезе коллагена. В зависимости от отдельной мутации, серьёзность синдрома может измениться от умеренного до опасного для жизни. Лечения нет, но существует терапия (уход), смягчающая последствия.

Синдром Робинова. Редкое генетическое заболевание, для которого на общем фоне отставания в развитии характерны — увеличенная голова, выпуклый лоб, низкий рост, полнота, брахидактилия конечностей, неразвитость внешних половых органов и др. Впервые синдром был описан в 1969 году в американском журнале заболеваний у детей педиатром и генетиком Мейнхардом Робиновым вместе с коллегами Фредериком Сильверманом и Хьюго Смитом. К 2002 году изучено и опубликовано в специализированной медицинской литературе более 100 клинических случаев заболевания. Синдром также известен как Робинов-Сильверман-Смит синдром. Рецессивная форма заболевания была ранее известна как синдром Covesdem.

Синдром Апе́ра (Аперта) — врожденная аномалия развития черепа, которая сочетается с отклонением развития кистей рук. Раннее закрытие венечного и стреловидного швов способствует деформации черепа, что приводит к внутричерепной гипертензии. Синдром Аперта является одной из форм акроцефалосиндактилии.

Глаз человека — парный сенсорный орган зрительной системы, обладающий способностью воспринимать электромагнитное излучение в световом диапазоне длин волн и обеспечивающий функцию зрения. Глаза расположены в передней части головы и вместе с веками, ресницами и бровями, являются важной частью лица. Область лица вокруг глаз активно участвует в мимике. Максимальный оптимум дневной чувствительности человеческого глаза приходится на максимум непрерывного спектра солнечного излучения, расположенный в «зелёной» области 550 (556) нм. При переходе от дневного освещения к сумеречному происходит перемещение максимума световой чувствительности по направлению к коротковолновой части спектра, и предметы красного цвета кажутся чёрными, синего (василёк) — очень светлыми.

Иссле́дование о́ргана зре́ния — исследование глаза человека посредством серии тестов, проводимых врачом-офтальмологом окулистом или ортоптистом с целью оценить остроту зрения, способность к зрительному сосредоточению, возможность различить объекты, а также другие исследования и измерения. Согласно мнению экспертов ВОЗ, все люди должны иметь доступ к периодическому и тщательному исследованию зрения в рамках плановых первичных медицинских осмотров, особенно после перенесённых травм или на фоне заболеваний глаз, ибо многие осложнения могут длительное время протекать бессимптомно. Кроме того, профилактический осмотр врачом-специалистом может выявить заболевания глаз, которые без своевременного лечения могут прогрессировать и стать причиной развития слепоты, а также глазные проявления многих системных заболеваний или признаки наличия новообразований или других аномалий головного мозга.

X-сцепленное рецессивное наследование — один из видов сцепленного с полом наследования. Такое наследование характерно для признаков, гены которых расположены на Х-хромосоме и которые проявляются только в гомозиготном или гемизиготном состоянии. Такой тип наследования имеет ряд врождённых наследственных заболеваний у человека, эти заболевания связаны с дефектом какого-либо из генов, расположенных на половой Х-хромосоме, и проявляются в случае, если нет другой Х-хромосомы с нормальной копией того же гена. В литературе встречается сокращение XR для обозначения X-сцепленного рецессивного наследования.

Пигментный ретинит (RP) — наследственное, дегенеративное заболевание глаз, которое вызывает сильное ухудшение зрения и часто слепоту. Прогрессирование RP не является последовательным. Некоторые люди могут иметь симптомы с детства, другие могут заметить симптомы позднее. В общем, чем позже начало, тем быстрее ухудшение зрения. Те, кто не имеют RP имеют 90-градусное периферийное зрение, в то время как люди, имеющие RP имеют периферийное зрение менее 90 градусов.

PRP31 — обрабатывающий пре-мРНК фактор 31, гомолог (англ. «PRP31 pre-mRNA processing factor 31 homolog »), также известный как PRPF31 , — белок, кодируемый у человека геном PRPF31.
Зрительный нерв содержит аксоны нервных клеток, которые образуют сетчатку, покидают глаз сквозь диск зрительного нерва, и следуют к зрительной коре, где входящие сигналы от глаз перерабатывается в изображение. Всего существует 1,2 миллиона волокон зрительного нерва, которые выходят из ганглионарных клеток внутренней сетчатки. Оптическая нейропатия — повреждение зрительного нерва по какой-либо причине. Повреждение и гибель этих нервных клеток, или нейронов, приводит к характерных особенностям оптической нейропатии. Основным симптомом является потеря цветового зрения. При медицинское обследовании зрительного нерва, оптическая нейропатия может быть визуализирована с помощью офтальмоскопа. Бледный диск характерен для давней оптической невропатии. Во многих случаях только один глаз влияет и пациенты могут не знать о потере цветового зрения, пока врач не попросит их закрыть здоровый глаз.
Доминантная атрофия зрительного нерва , или доминантная атрофия зрительного нерва типа Кьер — аутосомное наследственное заболевание, которое влияет на зрительный нерв, что приводит к снижению остроты зрения и слепоте, начиная с детства. Это обусловлено митохондриальной дисфункцией, опосредующей смерть волокон зрительного нерва. Доминантная атрофия зрительного нерва была впервые описана клинически Баттеном в 1896 году и названа оптической нейропатией Кьера в 1959 году в честь датского офтальмолога Poul Kjer, который исследовал 19 семей с болезнью. Хотя домининантная атрофия зрительного нерва является наиболее распространённой аутосомной наследственной оптической нейропатией и не связана с глаукомой, последняя часто ошибочно диагностируется вместо Кьер.
Друзы оптического диска (ODD) или друзы глазного нерва (ONHD) —глобулы из мукопротеинов и мукополисахаридов, которые постепенно кальцифицируют диск зрительного нерва. Они могут стать остатками транспортной системы аксонов вырождающихся ганглиозных клеток сетчатки. ODD также приписывались такие проблемы как врождённые поднятые или аномальные диски, псевдопапиллоэдема, псевдоневрит, утопление друз диска, и гиалиновые тельца в оптическом диске. Они могут быть связаны с потерей зрения различной степени и иногда приводить к слепоте, или, чаще, иметь бессимптомное течение.
Синдро́м Ке́рнса — Се́йра (англ. Kearns–Sayre syndrome, сокращённо KSS) — митохондриальная миопатия с типичным началом до 20-летнего возраста. KSS является более серьёзным синдромным вариантом хронической прогрессирующей внешней офтальмоплегии, синдром, который характеризуется изолированным поражением мышц, контролирующих движения век и контролирующих движения глаз. Это приводит к птозу и офтальмоплегии соответственно. KSS включает в себя триаду уже описанных CPEO, двустороннюю пигментную ретинопатию и блокаду сердца. Другие области участия могут включать в себя мозжечковую атаксию, проксимальную мышечную слабость, глухоту, сахарный диабет, дефицит гормона роста, гипопаратиреоз или другие эндокринные нарушения. В обоих этих заболеваниях вовлечение мышц может начаться односторонним, но всегда развивается в двусторонний дефицит, который является прогрессирующим.

Зрительный тракт — пучок нервных волокон, начинающийся от зрительного перекреста и заканчивающийся в подкорковых центрах зрительного анализатора — латеральном коленчатом теле, подушке таламуса и верхнем холмике крыши среднего мозга; входит в состав проводящего пути зрительного анализатора.
Синдром Пирсона — тяжелое врожденное аутосомно-рецессивное заболевание, вызванное мутацией гена, ответственного за кодирование β2-цепи ламинина. На долю которого приходится около 2,5% нефротического синдрома в течение первого года жизни, обычно клинически проявляющегося в течение первых 3 месяцев. В России зафиксирован единичный случай этого заболевания.
Синдро́м Ло́уренса-Му́на. Редкое аутосомно-рецессивное генетическое заболевание, характеризующиеся ожирением, умственной отсталостью, атаксией, дистрофией сетчатки, гипопитуитаризмом, большим количеством пальцев рук или ног. Синдром связан с нарушением функции гипоталамических центров.