Пороки развития — совокупность отклонений от нормального строения организма, возникающих в процессе внутриутробного или, реже, послеродового развития.

Транспозиция внутренних органов — редкий вариант биологической аномалии, в котором основные внутренние органы имеют зеркальное расположение по сравнению с обычным нормальным положением: верхушка сердца обращена вправо, печень расположена слева, желудок — справа. Обычное нормальное расположение органов на латыни называется situs solitus. В редких случаях встречается неопределённое положение внутренних органов, которое называется гетеротаксия, или situs ambiguous.

Врождённый поро́к се́рдца (ВПС) — дефект в структуре сердца и (или) крупных сосудов, присутствующий с рождения. Большинство пороков нарушают ток крови внутри сердца или по большому (БКК) и малому (МКК) кругам кровообращения. Пороки сердца являются наиболее частыми врождёнными дефектами и являются основной причиной детской смертности от пороков развития.

Синдром Вольфа — Хиршхорна — редкое генетическое заболевание, возникающее в результате делеции короткого плеча 4-й хромосомы. Впервые описан в 1965 году.

Аномалия Эбштейна — редкий врождённый порок сердца. Данная патология встречается с частотой 1% от всех врождённых пороков развития сердца. Впервые описана в 1866 году патологоанатомом В. Эбштейном. При этом пороке сердца створки правого атриовентрикулярного клапана исходят из стенок правого желудочка, а не из предсердно-желудочкового кольца и неполностью смыкаются. Таким образом, полость правого желудочка оказывается уменьшённой по сравнению с нормой, а часть правого желудочка от предсердно-желудочкового кольца до смещённого вниз трехстворчатого клапана становится продолжением правого предсердия. Также при аномалии наблюдается незаращение овального отверстия. Из-за таких анатомических изменений аномалия Эбштейна характеризуется
- Недостаточностью трёхстворчатого клапана.
- Незаращённым овальным отверстием и
- Уменьшением полости правого желудочка. Правое предсердие увеличивается в размерах и расширяется. Часть венозной крови перетекает в левое предсердие через открытое овальное отверстие и смешивается с артериальной. Это приводит к уменьшению содержания кислорода в артериальной крови и гипоксии органов и тканей. Возможны нарушения ритма сердца.
Атрезия — врождённое отсутствие или приобретенное заращение естественных отверстий и каналов в организме.

Коарктация аорты — врождённый порок сердца, проявляющийся сегментарным сужением просвета аорты. Лечение коарктации аорты хирургическое.
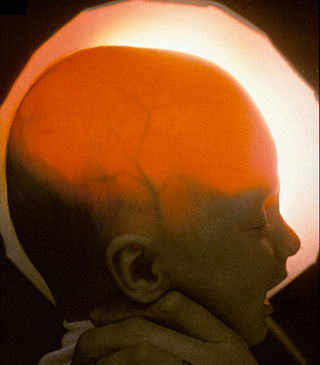
Гидранэнцефалия — крайне редкая, врождённая аномалия головного мозга, характеризующиеся полным или почти полным отсутствием больших полушарий, которые замещены полостью, заполненной спинномозговой жидкостью. Частота этого редкого порока развития составляет около одного случая на 18000 новорождённых. У больных отсутствует кость черепа. Продолговатый мозг и мозжечок сохранены. Наблюдаются судороги, грубые моторные и психические нарушения. Эффективного лечения этой аномалии развития не существует, поэтому, как правило, дети умирают в течение первого года жизни. Первый год жизни — наиболее труден для детей с этим пороком развития, но выживание всё-таки возможно. Известен случай, когда больной дожил до 32 лет.

Синдро́м Шереше́вского — Тёрнера (СШТ) — хромосомный симптомокомплекс, обусловленный частичной или полной моносомией по X-хромосоме, в то время как здоровые женщины имеют и унаследованную от матери хромосому, и хромосому от отца. Характеризуется аномалиями физического развития, низкорослостью, половым инфантилизмом и другими разнящимися признаками. Традиционным методом верифицирования является кариотипирование. Терапия основана на приёме гормонов роста и эстрогенов.
Синдро́м Ка́ллмана — симптомокомплекс наследственно обусловленных аномалий, характеризующийся сочетанием гипогонадизма с расстройствами обоняния и недостаточной секрецией гонадотропин-рилизинг гормона (ГнРГ). Низкий уровень ЛГ и ФСГ ведёт к развитию вторичного (центрального) гипогонадизма.

Синдро́м Нуна́н — редкая врождённая патология, как правило, наследуется по аутосомно-доминантному типу, носит семейный характер, однако встречаются и спорадически. Синдром предполагает наличие фенотипа, характерного для синдрома Шерешевского-Тернера у особей женского и мужского пола с нормальным генотипом.

Синдром кошачьего глаза, также Синдром кошачьих зрачков, Синдром Шмида-Фраккаро — это редкая врождённая геномная патология, вызванная присутствием в кариотипе пациента маленькой дополнительной хромосомы, состоящей из материала 22-й хромосомы. Название синдрома «кошачий глаз» связано с тем, что одной из клинических манифестаций этого синдрома является вертикальная колобома глаза. Следует отметить, что около половины пациентов не имеют этой черты, так как для данного синдрома характерна крайне высокая вариабельность клинических проявлений, наблюдающаяся даже в случае семейной формы этого синдрома. Среди пациентов, у которых отсутствуют аномалии, угрожающие жизни, не наблюдается значительного уменьшения продолжительности жизни.
Гипертропия — смещения глаз (косоглазие), в результате чего визуального ось одного глаза направлена выше точки фиксации взгляда. Гипотропия — аналогичное условие, когда визуальная ось направлена ниже точки фиксации взгляда. Диссоциация вертикального отклонения является особым типом гипертропии, ведущей к медленному дрейфу вверх одного или редко обоих глаз, как правило, когда пациент невнимателен.

Дуга аорты — часть аорты, самого крупного артериального сосуда в теле человека. Начинается на уровне хряща II ребра справа от расположенного внутриперикардиально восходящего отдела аорты и на уровне тел III—IV грудных позвонков переходит в нисходящий отдел.

Пентасомия по X-хромосоме (49,XXXXX) — наследственное нарушение, обусловленное наличием дополнительных X хромосом, является частным случаем анеуплоидии. Представляет собой хромосомную аномалию, при которой у женщины пять Х-хромосом вместо двух. Признаки могут включать умственную отсталость, низкий рост, низко посаженные уши, мышечную гипотонию и задержку развития. Осложнения могут включать врожденные пороки сердца.
Синдром 49, XXXXY — чрезвычайно редкая анеуплоидная половая хромосомная аномалия. Случается примерно в 1 из 85 000 до 100 000 случаев. Этот синдром является результатом материнского нерасхождения хромосом во время мейоза I и II. Впервые он был диагностирован в 1960 году и был назван синдромом Фраккаро по имени исследователя.
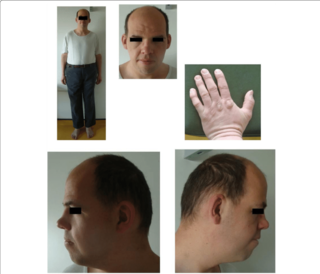
Синдром 49, XYYYY — чрезвычайно редкая анеуплоидная половая хромосомная аномалия. Случается реже чем 1 из 1 000 000 случаев.
Синдром 48, XYYY — чрезвычайно редкая анеуплоидная половая хромосомная аномалия. Случается реже чем 1 из 1 000 000 случаев.

Агенези́я мозо́листого те́ла (АМТ) — редкий врожденный дефект, при котором наблюдается полное или частичное отсутствие мозолистого тела. АМТ может быть обнаружена изолированно на МРТ, но чаще сочетается с множественными пороками развития.
Синдром микроделеции 1p31p32 — редкая хромосомная аномалия. Встречается реже, чем 1 на 1 000 000 случаев. Представляет собой частичную утрату участка 31p32 короткого плеча хромосомы 1 с переменным фенотипом, в основном характеризующимся задержкой развития, агенезией/гипоплазией мозолистого тела и характерными чертами лица, такими как макроцефалия, микрогнатией, низко посаженными ушами, загнутыми вперед ноздрями и т. д. Также часто наблюдаются дефекты мочевыводящих путей. Другие проблемы включают в себя судороги, гипотонию, нарушение функции спинного мозга и порок Киари I типа.