
Синдро́м нечувстви́тельности к андроге́нам — врожденные эндокринные вариации полового развития, вызванные мутацией гена, отвечающего за андрогеновый рецептор. Подобные синдромы варьируются в зависимости от структуры и чувствительности аномального рецептора. Клинические фенотипы находятся в диапазоне от номинального мужского телосложения с мягким синдромом сперматогенеза до крайне феминного телосложения, в дополнение к наличию Y-хромосомы.
Мозаицизм — от фр. mosaique «мозаика» — наличие в тканях генетически различающихся клеток.

Синдром Э́двардса — хромосомное заболевание, характеризуется комплексом множественных пороков развития и трисомией 18 хромосомы. Описан в 1960 году Джоном Эдвардсом. Популяционная частота примерно 1:3000 в США и 1:5000 в мире на 2016 год. Дети с трисомией в 18-й хромосоме чаще рождаются у пожилых матерей, взаимосвязь с возрастом матери менее выражена, чем в случаях трисомии хромосомы 21 и 13. Для женщин старше 45 лет риск родить больного ребёнка составляет 0,7 %. Девочки с синдромом Эдвардса рождаются в три раза чаще мальчиков. Выживание после года жизни составляет около 5—10 %.

Синдро́м Да́уна — хромосомная болезнь, чаще всего вызванная тем, что хромосомы 21-й пары представлены тремя копиями вместо нормальных двух. Существует ещё две формы этого синдрома: транслокация хромосомы 21 на другие хромосомы — 4 % случаев, и мозаичный вариант синдрома — 5 %.
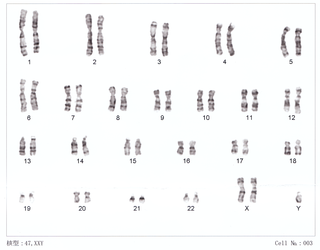
Синдром Клайнфельтера — генетическое отклонение. Клиническая картина синдрома описана в 1942 году в работах Гарри Клайнфельтера и Фуллера Олбрайта. Генетической особенностью этого синдрома является разнообразие цитогенетических вариантов и их сочетаний (мозаицизм). Обнаружено несколько типов полисомии по хромосомам X и Y у лиц мужского пола: 47, XXY; 47, XYY; 48, XXXY; 48, XYYY; 48 XXYY; 49 XXXXY; 49 XXXYY. Наиболее распространён синдром Клайнфельтера. Общая частота его колеблется в пределах 1 на 500—700 новорождённых мальчиков, что делает данный синдром первым по частоте встречаемости среди хромосомных болезней.

Синдро́м Шереше́вского — Тёрнера (СШТ) — хромосомный симптомокомплекс, обусловленный частичной или полной моносомией по X-хромосоме, в то время как здоровые женщины имеют и унаследованную от матери хромосому, и хромосому от отца. Характеризуется аномалиями физического развития, низкорослостью, половым инфантилизмом и другими разнящимися признаками. Традиционным методом верифицирования является кариотипирование. Терапия основана на приёме гормонов роста и эстрогенов.
Анеуплоиди́я — изменение кариотипа, при котором число хромосом в клетках не кратно гаплоидному набору (n). Отсутствие в хромосомном наборе диплоидного организма одной хромосомы называется моносомией (2n-1); отсутствие двух гомологичных хромосом — нуллисомией (2n-2); наличие дополнительной хромосомы называется трисомией (2n+1). Анеуплоидия возникает в результате нарушения сегрегации хромосом в митозе или мейозе. Анеуплоидия вызывает у человека некоторые наследственные синдромы. Анеуплоидия по аутосомам нарушает нормальное эмбриональное развитие и является одной из основных причин спонтанных абортов. Анеуплоидия характерна для опухолевых клеток, особенно для клеток сóлидных опухолей. Патологический фенотип при анеуплоидии формируется из-за нарушения дозового баланса генов, при моносомии дополнительный негативный вклад оказывает гемизиготное состояние генов моносомной хромосомы.

Кариоти́п — совокупность признаков полного набора хромосом, присущая клеткам данного биологического вида, данного организма или линии (клона) клеток. Графическое изображение кариотипа, то есть набора хромосом при расположении их по группам в зависимости от формы и величины, называют — идиограмма (кариограмма). Не путать с идеограммой.
Синдром Свайера, XY дисгенезия гонад, женская гонадальная дисгенезия или гонадальная дисгенезия — генетическое нарушение, вариант гипогонадизма с кариотипом 46,XY. Организм человека с синдромом Свайера имеет характерный для мужского организма набор хромосом, но половые железы представляют собой гонадный тяж и не производят гормоны. В результате он имеет женские гениталии, женскую репродуктивную систему и выглядит как женщина. В период полового созревания развитие вторичных половых признаков не происходит и наблюдается аменорея. Существует практика удаления гонад в раннем возрасте с целью предотвращения развития рака.
Синдро́м де ля Шапе́ля — назван в честь исследователя, впервые охарактеризовавшего его в 1972 году. Синдром относится к редкой хромосомной патологии, возникающей в результате кроссинговера между X- и Y-хромосомами в процессе мейоза, в результате чего одна или обе X-хромосомы содержат нормальный мужской ген SRY. Распространённость данного синдрома составляет 4—5 на 100 000, что меньше встречаемости синдрома Клайнфельтера.

XYY-синдром, также известен как YY-синдром или Синдром Джейкобс, — хромосомное заболевание, характерное только для биологических мужчин. Носитель синдрома имеет дополнительную Y-хромосому, общий хромосомный набор составляет 44 аутосомы и три половые хромосомы. Внешне мужчины с дополнительной Y-хромосомой обычно не имеют существенных отличий от мужчин с обычным набором хромосом, но могут иметь ряд особенностей.

Синдром кошачьего глаза, также Синдром кошачьих зрачков, Синдром Шмида-Фраккаро — это редкая врождённая геномная патология, вызванная присутствием в кариотипе пациента маленькой дополнительной хромосомы, состоящей из материала 22-й хромосомы. Название синдрома «кошачий глаз» связано с тем, что одной из клинических манифестаций этого синдрома является вертикальная колобома глаза. Следует отметить, что около половины пациентов не имеют этой черты, так как для данного синдрома характерна крайне высокая вариабельность клинических проявлений, наблюдающаяся даже в случае семейной формы этого синдрома. Среди пациентов, у которых отсутствуют аномалии, угрожающие жизни, не наблюдается значительного уменьшения продолжительности жизни.

Микрохромосо́мы — очень маленькие хромосомы, типичные для кариотипов птиц, некоторых рептилий, рыб и земноводных; у млекопитающих они, судя по всему, отсутствуют. Их размер составляет меньше 20 мегабаз; хромосомы, чей размер превышает 40 мегабаз, называются макрохромосомами, а хромосомы размером от 20 до 40 мегабаз — промежуточными хромосомами.

Пентасомия по X-хромосоме (49,XXXXX) — наследственное нарушение, обусловленное наличием дополнительных X хромосом, является частным случаем анеуплоидии. Представляет собой хромосомную аномалию, при которой у женщины пять Х-хромосом вместо двух. Признаки могут включать умственную отсталость, низкий рост, низко посаженные уши, мышечную гипотонию и задержку развития. Осложнения могут включать врожденные пороки сердца.
Синдром 49, XXXXY — чрезвычайно редкая анеуплоидная половая хромосомная аномалия. Случается примерно в 1 из 85 000 до 100 000 случаев. Этот синдром является результатом материнского нерасхождения хромосом во время мейоза I и II. Впервые он был диагностирован в 1960 году и был назван синдромом Фраккаро по имени исследователя.

Синдром 48, XXYY — это аномалия хромосом, при которой у человека имеется дополнительная X и Y-хромосома. Клетки человека обычно содержат две половые хромосомы, одну от матери и одну от отца. Обычно женщины имеют две Х-хромосомы (XX), а мужчины имеют одну Х и одну Y-хромосому (XY). Появление по крайней мере одной Y-хромосомы с правильно функционирующим геном SRY делает человека физиологически мужчиной. Следовательно, люди с XXYY являются генотипически мужчинами. Мужчины с синдромом XXYY имеют 48 хромосом вместо типичных 46. По оценкам, он случается в 1 случае на каждые 18 000-40 000 новорожденных.
Смешанная дисгенезия гонад, также известный как X0/XY-мозаицизм и 45,X/46,XY-мозаицизм — редкий синдром, связанный с анеуплоидией половых хромосом и мозаицизмом Y-хромосомы. Также называется мозаичным кариотипом.

Истинный гермафродитизм, также известный как овотестикулярное расстройство полового развития, является медицинским термином, обозначающим феномен при котором человек рождается с тканями яичника и яичка. Чаще всего одна или обе гонады — это овотестис, содержащий оба типа ткани.
Синдром WAGR – это редкий наследственный аутосомно-доминантный синдром (частота 1:500000—1:1000000 человек, который клинически характеризуется сочетанием опухоли Вильмса, аниридии, урогенитальных аномалий и/или гонадобластомы и задержки психомоторного развития. Синдром встречается примерно в 8 % случаев врожденной аниридии, и в 0,75% всех случаев опухоли Вильмса.
Однородительская дисомия — это явление, которое происходит, когда человек получает две копии хромосомы или части хромосомы от одного родителя и ни одной копии от другого родителя. Однородительская дисомия (ОД) может быть результатом гетеродисомии, при которой пара неидентичных хромосом наследуется от одного родителя, или изодисомии, при которой дублируется одна хромосома от одного родителя. Однородительская дисомия может иметь клиническое значение по нескольким причинам. Например, изодисомия или гетеродисомия могут нарушить специфичный для родителей геномный импринтинг, что приведет к нарушениям импринтинга. Кроме того, изодисомия приводит к большим блокам гомозиготности, что может привести к обнаружению рецессивных генов, аналогичное явление наблюдается у инбредных детей единокровных партнёров. Было обнаружено, что ОД встречается примерно в 1 из 2000 родов.
