
Акрокефа́лия — аномальная вытянутая башнеобразная форма головы, вызываемая преждевременным зарастанием венечного шва. Иногда акрокефалией называют преобладание высоты черепа над шириной при нормальном строении швов.

Синдром (болезнь) Марфана аутосомно-доминантное заболевание из группы наследственных патологий соединительной ткани. Синдром вызван мутациями генов, кодирующих синтез гликопротеина фибриллина-1, и является плейотропным. Заболевание характеризуется различной пенетрантностью и экспрессивностью. В классических случаях лица с синдромом Марфана высоки (долихостеномелия), имеют удлинённые конечности, вытянутые пальцы (арахнодактилия) и недоразвитие жировой клетчатки. Помимо характерных изменений в органах опорно-двигательного аппарата, наблюдается патология в органах зрения и сердечно-сосудистой системы, что в классических вариантах составляет триаду Марфана.
Гипофосфатазия (ГФФ) — редкое (орфанное), жизнеугрожающее, генетически обусловленное, врождённое заболевание, проявляющееся нарушением минерализации костей скелета и зубов, а также системными осложнениями, включая нарушение дыхания, судороги, мышечную слабость, боль в костях и нефрокальциноз.
Синдро́м (болезнь) де То́ни — Дебре́ — Фанко́ни — врождённое заболевание, наследуется по аутосомно-рецессивному типу. Комплекс биохимических и клинических проявлений поражения проксимальных почечных канальцев с нарушением канальцевой реабсорбции фосфата, глюкозы, аминокислот и бикарбоната. Одно из рахитоподобных заболеваний.

Синдром Ваарденбурга — наследственное заболевание. Имеет следующие клинические признаки: телекант, гетерохромия радужки, седая прядь надо лбом и врождённая тугоухость различной степени.
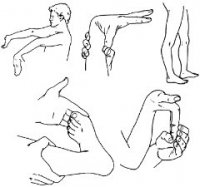
Синдром Э́лерса — Данло́са — это группа наследственных системных дисфункций соединительной ткани, вызванных дефектом в синтезе коллагена. В зависимости от отдельной мутации, серьёзность синдрома может измениться от умеренного до опасного для жизни. Лечения нет, но существует терапия (уход), смягчающая последствия.

Дисплазия соединительной ткани — системное заболевание соединительной ткани, генетически гетерогенное и клинически полиморфное патологическое состояние, обусловленное нарушением развития соединительной ткани в эмбриональном и постнатальном периодах. Характеризуется дефектами волокнистых структур и основного вещества соединительной ткани, приводящее к расстройству гомеостаза на тканевом, органном и организменном уровнях в виде различных морфофункциональных нарушений висцеральных и локомоторных органов с прогредиентным течением. Синонимы: соединительнотканная дисплазия, наследственное нарушение соединительной ткани, врожденная соединительнотканная недостаточность, системное невоспалительное заболевание соединительной ткани, гипермобильный синдром, наследственная коллагенопатия.

Синдром Робинова. Редкое генетическое заболевание, для которого на общем фоне отставания в развитии характерны — увеличенная голова, выпуклый лоб, низкий рост, полнота, брахидактилия конечностей, неразвитость внешних половых органов и др. Впервые синдром был описан в 1969 году в американском журнале заболеваний у детей педиатром и генетиком Мейнхардом Робиновым вместе с коллегами Фредериком Сильверманом и Хьюго Смитом. К 2002 году изучено и опубликовано в специализированной медицинской литературе более 100 клинических случаев заболевания. Синдром также известен как Робинов-Сильверман-Смит синдром. Рецессивная форма заболевания была ранее известна как синдром Covesdem.

Краниосиностоз — раннее закрытие черепных швов, что способствует ограниченному объему черепа, его деформации и внутричерепной гипертензии. Заболевание встречается у одного новорождённого на 2000, чаще наблюдается у мальчиков.
Синдро́м Клиппе́ля — Фейля — редкий врожденный порок развития шейных и верхнегрудных позвонков, который характеризуется наличием у больного короткой и малоподвижной шеи. Шейные позвонки при этом срощены в единый массив. Это наследственная патология, она передаётся по аутосомно-доминантному типу. Впервые заболевание было описано французскими врачами Морисом Клиппелем и Андре Фейлем в 1912 году.

Синдром Тричера Коллинза — аутосомно-доминантное заболевание, характеризующееся черепно-лицевой деформацией. Описан английским офтальмологом Эдвардом Тричером Коллинзом в 1900 году.

Брахидактилия — аномалия развития рук или ног, укорочение пальцев. Брахидактилия часто может сочетаться с симфалангизмом и с различными формами синдактилии. Брахидактилия наследуется по аутосомно-доминантному типу.

Синдро́м Нуна́н — редкая врождённая патология, как правило, наследуется по аутосомно-доминантному типу, носит семейный характер, однако встречаются и спорадически. Синдром предполагает наличие фенотипа, характерного для синдрома Шерешевского-Тернера у особей женского и мужского пола с нормальным генотипом.

X-сцепленное рецессивное наследование — один из видов сцепленного с полом наследования. Такое наследование характерно для признаков, гены которых расположены на Х-хромосоме и которые проявляются только в гомозиготном или гемизиготном состоянии. Такой тип наследования имеет ряд врождённых наследственных заболеваний у человека, эти заболевания связаны с дефектом какого-либо из генов, расположенных на половой Х-хромосоме, и проявляются в случае, если нет другой Х-хромосомы с нормальной копией того же гена. В литературе встречается сокращение XR для обозначения X-сцепленного рецессивного наследования.
Синдро́м Гу́рлер — тяжёлое наследственное заболевание из группы мукополисахаридозов, относящихся к лизосомным болезням накопления. Характеризуется недостаточностью альфа-L-идуронидазы — фермента лизосом, участвующего в катаболизме кислых мукополисахаридов, которые составляют основу межклеточного вещества соединительной ткани.

Мукополисахаридо́з I — группа метаболических заболеваний соединительной ткани, связанных с нарушением обмена кислых гликозаминогликанов, вызванных недостаточностью лизосомного фермента обмена гликозаминогликанов альфа-L-идуронидазы. Данная группа мукополисахаридозов связана с наследственной аномалией, вызванной генетически детерминированным дефектом гена, расположенного в локусе 4q16.3, которая наследуется по аутосомно-рецессивному типу. Проявляется в виде лизосомной болезни накопления, которая приводит к различным дефектам нервной, костной, хрящевой и соединительной тканей.
Синдром Марото — Лами — редкая наследственная болезнь, одна из форм мукополисахаридоза из группы лизосомных болезней накопления, биохимически связанная с дефицитом фермента лизосом N-Ацетилгексозамин-4-сульфатсульфатазы.

Синдром Морганьи — Стюарта — Мореля — состояние, связанное с широким спектром эндокринных проблем, включая сахарный и несахарный диабеты и гиперпаратиреоз. Другие признаки и симптомы включают головную боль, головокружение, избыточное оволосение по мужскому типу, проблемы с менструацией, галакторею, ожирение, депрессию и эпилептические припадки. Утолщение внутренней поверхности лобной части черепа является, как правило, начальной стадией заболевания, известного как hyperostosis frontalis interna.
Синдро́м Ло́уренса-Му́на. Редкое аутосомно-рецессивное генетическое заболевание, характеризующиеся ожирением, умственной отсталостью, атаксией, дистрофией сетчатки, гипопитуитаризмом, большим количеством пальцев рук или ног. Синдром связан с нарушением функции гипоталамических центров.

Гипофосфатемический рахит, Х-ГФР, фосфат-диабет, витамин-Д-резистентный рахит — это генетически гетерогенная группа заболеваний, характеризующаяся развитием рахита из-за повышенного выведения фосфора из организма по причине нарушения реабсорбции фосфатов в почках, то есть обратного всасывания фосфора из первичной мочи в кровь. Средний возраст манифестации от 6 месяцев до 1,5 лет. В связи с тем, что фосфор необходим для нормального роста и развития костей, дети с этим заболеванием часто имеют рост ниже среднего и деформации нижних конечностей.

