
Синдро́м Туре́тта — генетически обусловленное расстройство центральной нервной системы, которое проявляется в любом возрасте и характеризуется множественными двигательными («моторными») тиками и как минимум одним голосовым, появляющимися много раз в течение дня. В американском Диагностическом и статистическом руководстве по психическим расстройствам пятого издания (DSM-5), наряду с остальными тикозными расстройствами, относится к нейроонтогенетическим моторным расстройствам. Довольно часто встречается коморбидное состояние: синдром Туретта с синдромом дефицита внимания и гиперактивности.

Мультиформная глиобласто́ма — наиболее частая и наиболее агрессивная форма опухоли мозга, которая составляет до 52 % первичных опухолей мозга и до 20 % всех внутричерепных опухолей. Несмотря на то, что глиобластома является наиболее частой первичной опухолью мозга, на 100 000 жителей Европы и Северной Америки регистрируется всего 2—3 случая заболевания. Термин «глиобластома» подразумевает два варианта этой болезни: гигантоклеточная глиобластома и глиосаркома.

Тератома — опухоль, образующаяся из гоноцитов, наиболее часто в яичниках у женщин, в яичках у мужчин, в крестцово-копчиковой области у детей, а также в мозге. Представляет собой ткань или даже орган, нетипичный для локализации опухоли: в тератоме могут присутствовать волосы, мышечная ткань, костная ткань, реже более сложные органы — глаз, туловище, конечности.
Синдром Фрейзера — аутосомно-рецессивное врождённое заболевание. Синдром назван по имени медицинского генетика Джорджа Фрейзера.

Инсулино́ма — доброкачественное новообразование, бесконтрольно секретирующее в кровяное русло инсулин, что приводит к развитию гипогликемического симптомокомплекса и чаще проявляется тощаковым гипогликемическим синдромом. Гораздо реже встречаются инсулинсекретирующие APUDомы (апудомы) — опухоли из параэндокринных клеток, локализацию которых установить крайне сложно. Имеются сообщения об инсулиномах, возникающих из энтерохромаффинных клеток кишечника. Злокачественные инсулиномы составляют 10—15 %, треть из которых метастазирует. У 4—14 % больных инсулиномы множественные, около 2 % новообразований располагаются вне поджелудочной железы. Инсулинсекретирующая опухоль описана во всех возрастных группах — от новорожденых до престарелых, тем не менее чаще она проявляется в наиболее трудоспособном возрасте — от 30 до 55 лет. Среди общего числа больных дети составляют около 5 %.

Множественная эндокринная неоплазия типа IIb — в данный синдром вошли те же опухоли, что и в МЭН IIa, однако первичный гиперпаратиреоз проявляется реже, а медуллярная карцинома щитовидной железы протекает крайне злокачественно. Выявляется в возрасте 4—37 лет. Отличается от МЭН IIa наличием нейрином (невром) слизистых оболочек и расстройств опорно-двигательного аппарата. Нейриномы чаще локализуются на губах, щеках, языке, других отделах желудочно-кишечного тракта и представляют собой бело-розовые безболезненные узелки диаметром от 1—3 мм до 1 см. Множественная эндокринная неоплазия типа IIb является самым серьёзным вариантом синдрома МЭН.

Синдро́м Ба́рттера — форма гиперальдостеронизма с гиперплазией юкстагломерулярного аппарата почек и резистентностью к сосудосуживающему действию ангиотензина II, обусловленной внешними (вторичными) нарушениями передачи сигнала ангиотензина II.
Синдро́м де ля Шапе́ля — назван в честь исследователя, впервые охарактеризовавшего его в 1972 году. Синдром относится к редкой хромосомной патологии, возникающей в результате кроссинговера между X- и Y-хромосомами в процессе мейоза, в результате чего одна или обе X-хромосомы содержат нормальный мужской ген SRY. Распространённость данного синдрома составляет 4—5 на 100 000, что меньше встречаемости синдрома Клайнфельтера.

Синдром Проте́я — очень редкое врождённое заболевание, характеризующееся чрезмерно быстрым и атипичным ростом кости, а также кожных покровов. Часто сопровождается опухолями отдельных частей тела.

BTLA, или B- и T-лимфоцитарный аттенюатор, — мембранный белок, лимфоцитарный ингибирующий рецептор, участвует в регуляции лимфоцитов во время иммунного ответа. Продукт гена человека BTLA.
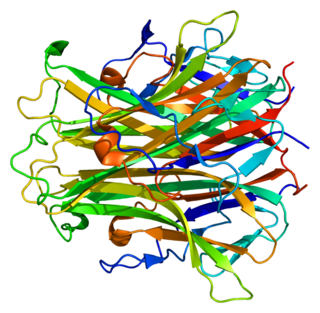
TNFSF11, или RANKL — мембранный белок, цитокин семейства факторов некроза опухоли. Продукт гена человека TNFSF11. Играет важную роль в метаболизме костной ткани, активируя остеокласты.
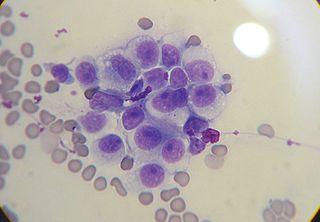
Трансмисси́вная венери́ческая о́пухоль соба́к — трансмиссивная гистиоцитная опухоль наружных половых органов собак и других псовых. Опухолевые клетки передаются между животными половым путём. Данная болезнь относится к категории трансмиссивных злокачественных опухолей, примером которых является лицевая опухоль тасманского дьявола.

TNFRSF1A — мембранный белок, рецептор из надсемейства рецепторов фактора некроза опухоли. У человека кодируется геном TNFRSF1A.

CD154, также лиганд CD40, CD40L, или TNFSF5 — цитокин семейства факторов некроза опухоли, в основном, локализующийся на активированных T-лимфоцитах. Лиганд рецептора CD40, экспрессируемого на антигенпредставляющих клетках, а также B-лимфоцитах. Продукт гена человека CD40LG.
Синдром Ушера — сравнительно редкое генетическое заболевание, вызываемое мутацией одного из 10 генов, приводящее к врождённой нейросенсорной тугоухости и прогрессирующей потере зрения. Одна из основных причин слепоглухоты. В настоящее время неизлечим. Наследуется по аутосомно-рецессивному принципу.
Синдром полтора является редким офтальмопаретическим синдромом, характеризующимся «параличем сопряженного горизонтального взгляда в одном направлении и межъядерной офтальмоплегией в другом». Наиболее частым проявлением этого необычного синдрома является ограничение горизонтального движения глаз с абдукцией одного глаза, без горизонтального перемещения другого глаза. Нистагм также присутствует при отведённом глазе на стороне, противоположной поражённой. Конвергенция классически управляется черепным нервом III и его ядро предоставляется на двусторонней основе.

Синдром Дуэйна — врождённый редкий тип косоглазия, чаще всего характеризуется неспособностью глаза двигаться наружу. Синдром был впервые описан офтальмологами Якобом Штиллингом (1887) и Зигмундом Тюрком (1896), а впоследствии стал носить имя Александра Дуэйна, который осветил этот синдром более подробно в 1905 году.

Тройной синдром (ААА), также известный как синдром Ахалазия-Аддисонианизма-Алакримии или синдром Оллгрова , — редкий аутосомно-рецессивный врожденный порок. В большинстве случаев характеризуется отсутствием семейной истории о нём. Синдром был обнаружен Джереми Оллгроувом и его коллегами в 1978 году. ААА представляет аббревиатуру синдрома ахалазии-аддисонианизма-алакримии. Алакримия является обычно ранним проявлением. Это прогрессирующее заболевание, которое может длиться годами, чтобы развить полноценную клиническую картину.
Синдро́м ли́зиса о́пухоли — группа метаболических нарушений, возникающих как осложнение при лечении онкологических заболеваний. Вызывается гибелью большого количества клеток опухоли за короткий период времени, при которой их содержимое попадает в кровь. Чаще всего возникает при лечении лимфом и лейкозов. В онкологии и гематологии это потенциально смертельное осложнение, поэтому пациенты с риском его возникновения должны находиться под наблюдением во время всего периода лечения химиотерапией.
Синдром WAGR – это редкий наследственный аутосомно-доминантный синдром (частота 1:500000—1:1000000 человек, который клинически характеризуется сочетанием опухоли Вильмса, аниридии, урогенитальных аномалий и/или гонадобластомы и задержки психомоторного развития. Синдром встречается примерно в 8 % случаев врожденной аниридии, и в 0,75% всех случаев опухоли Вильмса.