Синдро́м (болезнь) де То́ни — Дебре́ — Фанко́ни — врождённое заболевание, наследуется по аутосомно-рецессивному типу. Комплекс биохимических и клинических проявлений поражения проксимальных почечных канальцев с нарушением канальцевой реабсорбции фосфата, глюкозы, аминокислот и бикарбоната. Одно из рахитоподобных заболеваний.

Синдром Ваарденбурга — наследственное заболевание. Имеет следующие клинические признаки: телекант, гетерохромия радужки, седая прядь надо лбом и врождённая тугоухость различной степени.
Болезнь Виллебранда — Диана — наследственное заболевание крови, характеризующееся возникновением эпизодических спонтанных кровотечений, которые схожи с кровотечениями при гемофилии.
Синдром Э́лерса — Данло́са — это группа наследственных системных дисфункций соединительной ткани, вызванных дефектом в синтезе коллагена. В зависимости от отдельной мутации, серьёзность синдрома может измениться от умеренного до опасного для жизни. Лечения нет, но существует терапия (уход), смягчающая последствия.

Дисплазия соединительной ткани — системное заболевание соединительной ткани, генетически гетерогенное и клинически полиморфное патологическое состояние, обусловленное нарушением развития соединительной ткани в эмбриональном и постнатальном периодах. Характеризуется дефектами волокнистых структур и основного вещества соединительной ткани, приводящее к расстройству гомеостаза на тканевом, органном и организменном уровнях в виде различных морфофункциональных нарушений висцеральных и локомоторных органов с прогредиентным течением. Синонимы: соединительнотканная дисплазия, наследственное нарушение соединительной ткани, врожденная соединительнотканная недостаточность, системное невоспалительное заболевание соединительной ткани, гипермобильный синдром, наследственная коллагенопатия.

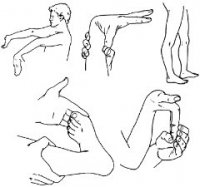
Синдром Апе́ра (Аперта) — врожденная аномалия развития черепа, которая сочетается с отклонением развития кистей рук. Раннее закрытие венечного и стреловидного швов способствует деформации черепа, что приводит к внутричерепной гипертензии. Синдром Аперта является одной из форм акроцефалосиндактилии.
Синдро́м Ка́ллмана — симптомокомплекс наследственно обусловленных аномалий, характеризующийся сочетанием гипогонадизма с расстройствами обоняния и недостаточной секрецией гонадотропин-рилизинг гормона (ГнРГ). Низкий уровень ЛГ и ФСГ ведёт к развитию вторичного (центрального) гипогонадизма.

Синдро́м Нуна́н — редкая врождённая патология, как правило, наследуется по аутосомно-доминантному типу, носит семейный характер, однако встречаются и спорадически. Синдром предполагает наличие фенотипа, характерного для синдрома Шерешевского-Тернера у особей женского и мужского пола с нормальным генотипом.

Синдро́м Рассе́ла-Си́львера — комплекс наследственных аномалий врождённый карликовый рост в результате нарушений эмбрионального развития: малая масса тела при рождении, низкий рост, задержка общего развития, треугольное лицо, опущенные вниз уголки рта, укороченные и согнутые пальцы рук, синдактилия.

Синдром Рубинштейна-Тейби впервые описан в 1963 г. J. Rubinstein и Н. Taybi.
Популяционная частота — 1:25 000 — 30000.
Соотношение полов — M1:Ж1.
Тип наследования неизвестен.
Первичные иммунодефициты — наследственные или приобретённые во внутриутробном периоде иммунодефицитные состояния. Обычно они проявляются или сразу после рождения, или в течение первых двух лет жизни. Однако менее выраженные генетические дефекты иммунного ответа могут манифестировать позже, например, на втором—третьем десятилетии жизни. Наследственные формы первичных иммунодефицитов, как правило, характеризуются аутосомно-рецессивным или рецессивным, сцепленным с Х-хромосомой типом наследования. Первым в 1952 г. был описан синдром Бру́тона.

Синдром Маркеза́ни — синдром микросферофакии-брахиморфии, врождённая гиперпластическая мезодермальная дисморфодистрофия. Данная патология характеризуется тремя основными признаками: низкий рост, подвывих хрусталика и брахидактилия. Также наблюдаются нарушение аккомодации, глаукома, брахицефалия и недоразвитие конечностей, ограничение подвижности в суставах, укорочение туловища и шеи.

X-сцепленное рецессивное наследование — один из видов сцепленного с полом наследования. Такое наследование характерно для признаков, гены которых расположены на Х-хромосоме и которые проявляются только в гомозиготном или гемизиготном состоянии. Такой тип наследования имеет ряд врождённых наследственных заболеваний у человека, эти заболевания связаны с дефектом какого-либо из генов, расположенных на половой Х-хромосоме, и проявляются в случае, если нет другой Х-хромосомы с нормальной копией того же гена. В литературе встречается сокращение XR для обозначения X-сцепленного рецессивного наследования.
Синдро́м Гу́рлер — тяжёлое наследственное заболевание из группы мукополисахаридозов, относящихся к лизосомным болезням накопления. Характеризуется недостаточностью альфа-L-идуронидазы — фермента лизосом, участвующего в катаболизме кислых мукополисахаридов, которые составляют основу межклеточного вещества соединительной ткани.
Синдро́м Шейе — тяжёлое наследственное заболевание из группы мукополисахаридозов, относящихся к лизосомным болезням накопления. Характеризуется недостаточностью альфа-L-идуронидазы — фермента лизосом, участвующего в катаболизме кислых мукополисахаридов, которые составляют основу межклеточного вещества соединительной ткани. Заболевание редкое, проявляется в детском возрасте.
GM1-ганглиозидо́зы — редкие наследственные заболевания из группы лизосомных болезней накопления. Развитие клинической картины обусловлено дефектом или недостатком β-галактозидазы, который ведёт к нарушению метаболизма и накоплению субстратов (ганглиозида GM1, гликопротеинов и кератансульфата) главным образом в нервных клетках центральной и периферической нервной системы.

Аутосо́мно-рецесси́вное насле́дование — свойственный диплоидным эукариотам тип наследования признака, контролируемого рецессивными аллелями аутосомного гена. Для проявления мутации или болезни с таким типом наследования мутантный аллель, локализованный в аутосоме, должен быть унаследован от обоих родителей. Иными словами, мутация проявляется только в гомозиготном состоянии, то есть тогда, когда обе копии гена, расположенные на гомологичных аутосомах, являются повреждёнными. Если мутация находится в гетерозиготном состоянии, и мутантному аллелю сопутствует нормальный функциональный аллель, то аутосомно-рецессивная мутация не проявляется.
Синдро́м Ло́уренса-Му́на. Редкое аутосомно-рецессивное генетическое заболевание, характеризующиеся ожирением, умственной отсталостью, атаксией, дистрофией сетчатки, гипопитуитаризмом, большим количеством пальцев рук или ног. Синдром связан с нарушением функции гипоталамических центров.
Насле́дственная остеодистрофи́я Олбра́йта — редко встречающееся состояние, характеризующееся разнообразием симптомов. Имеет аутосомно-доминантный тип наследования. Если нарушение передаётся со стороны матери, то оно характеризуется резистентностью к определённым гормонам, в первую очередь к паратиреоидному гормону (ПТГ). Такое состояние может быть, в зависимости от спектра мутации, псевдогипопаратиреозом типа 1A, 1B и 1C . При наследовании по отцовской линии остеодистрофия протекает без гормональных расстройств и называется псевдо-псевдогипопаратиреозом . Заболевание впервые описано американским эндокринологом Фуллером Олбрайтом с соавторами соответственно в 1942 и в 1952 годах. Псевдогипопаратиреоз типа 2 клинически сходен с другими типами заболевания, однако имеет аутосомно-рецессивный тип наследования. Характеризуется повышением уровня циклического аденозинмонофосфата (цАМФ) в ответ на введение экзогенного ПТГ.

Cиндром Ротмунда — Томсона — это наследственное аутосомно-рецессивное заболевание, проявляющееся пойкилодермией, ювенильной катарактой, гипогонадизмом, а также аномалиями костной системы и зубов.

