
Насле́дственные заболева́ния — заболевания, возникновение и развитие которых связано с различными дефектами и нарушениями в наследственном аппарате клеток. В основе наследственных заболеваний лежат мутации: хромосомные, генные и митохондриальные. Наследственные заболевания могут быть обусловлены мутациями, передаваемыми в семьях по наследству, или мутациями, вновь возникшими в клетках зародышевой линии, в зиготе или на очень ранних этапах развития. Наследственные болезни многочисленны и разнообразны по проявлениям.

Альбинизм — наследственное заболевание, полное или почти полное отсутствие пигмента меланина или хлорофилла. Проявляется отсутствием нормальной вида окраски кожи, волос, шерсти, радужной и пигментной оболочек глаз, зелёных частей растений.

Боле́знь Тея — Са́кса (GM2 ганглиозидоз, ранняя детская амавротическая идиотия) — редкое наследственное заболевание с аутосомно-рецессивным типом наследования, поражающее центральную нервную систему (спинной и головной мозг, а также менингеальные оболочки). Относится к группе лизосомных болезней накопления. Названо в честь британского офтальмолога Уоррена Тея (англ. Warren Tay, 1843—1928) и американского невролога Бернарда Сакса (англ. Bernard Sachs, 1858—1944), которые впервые описали это заболевание независимо друг от друга в 1881 и 1887 годах соответственно.

Синдро́м Ма́ртина — Белл — наследственное заболевание.
Синдро́м (болезнь) де То́ни — Дебре́ — Фанко́ни — врождённое заболевание, наследуется по аутосомно-рецессивному типу. Комплекс биохимических и клинических проявлений поражения проксимальных почечных канальцев с нарушением канальцевой реабсорбции фосфата, глюкозы, аминокислот и бикарбоната. Одно из рахитоподобных заболеваний.
Болезнь Виллебранда — Диана — наследственное заболевание крови, характеризующееся возникновением эпизодических спонтанных кровотечений, которые схожи с кровотечениями при гемофилии.
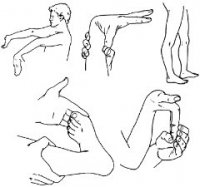
Синдром Э́лерса — Данло́са — это группа наследственных системных дисфункций соединительной ткани, вызванных дефектом в синтезе коллагена. В зависимости от отдельной мутации, серьёзность синдрома может измениться от умеренного до опасного для жизни. Лечения нет, но существует терапия (уход), смягчающая последствия.

Врождённая гиперплазия коры надпочечников (ВГКН) — группа заболеваний, наследуемых по аутосомно-рецессивному пути, при которых нарушается выработка кортизола надпочечниками. Гены, связанные с гиперплазией надпочечников, кодируют ферменты, участвующие в стероидогенезе — цепочке реакций по преобразованию холестерина в стероиды.

Синдром Робинова. Редкое генетическое заболевание, для которого на общем фоне отставания в развитии характерны — увеличенная голова, выпуклый лоб, низкий рост, полнота, брахидактилия конечностей, неразвитость внешних половых органов и др. Впервые синдром был описан в 1969 году в американском журнале заболеваний у детей педиатром и генетиком Мейнхардом Робиновым вместе с коллегами Фредериком Сильверманом и Хьюго Смитом. К 2002 году изучено и опубликовано в специализированной медицинской литературе более 100 клинических случаев заболевания. Синдром также известен как Робинов-Сильверман-Смит синдром. Рецессивная форма заболевания была ранее известна как синдром Covesdem.
Генные болезни — это большая группа заболеваний, возникающих в результате повреждения ДНК на уровне гена. Термин употребляется в отношении моногенных заболеваний, в отличие от более широкой группы — Наследственные заболевания (см.)
Анеми́я Фанко́ни — редкое наследственное заболевание. Встречается с частотой 1 на 350 000 новорождённых. Чаще выявляется среди евреев-ашкеназов и у народов Южной Африки.
Первичные иммунодефициты — наследственные или приобретённые во внутриутробном периоде иммунодефицитные состояния. Обычно они проявляются или сразу после рождения, или в течение первых двух лет жизни. Однако менее выраженные генетические дефекты иммунного ответа могут манифестировать позже, например, на втором—третьем десятилетии жизни. Наследственные формы первичных иммунодефицитов, как правило, характеризуются аутосомно-рецессивным или рецессивным, сцепленным с Х-хромосомой типом наследования. Первым в 1952 г. был описан синдром Бру́тона.

Синдро́м тетраамели́и — очень редкое врождённое наследственное заболевание, характеризующееся отсутствием четырёх конечностей. Другие части тела, такие как лицо, череп, репродуктивные органы, анус и таз, также подвержены порокам развития. Для синдрома тетраамелии характерно аутосомно-рецессивное наследование. Заболевание связано с мутацией в гене WNT3.
Болезнь Фарбера — очень редкая аутосомная рецессивная болезнь накопления лизосом. Ввиду дефицита фермента керамидазы накапливаются липиды, что приводит к нарушениям в суставах, печени, горле, центральной нервной системе и различных тканях. Керамидаза расщепляет жиры в клетках организма. В случае болезни Фарбера ген, ответственный за выработку этого фермента, изменён в результате мутации. Поэтому жиры расщепляются и накапливаются в разных частях тела. Появляются признаки этого расстройства.

Синдром Корнелии де Ланге — наследственное заболевание, проявляющееся умственной отсталостью и множественными аномалиями развития. Частота заболевания — примерно 1 на 10000.
Синдром Марото — Лами — редкая наследственная болезнь, одна из форм мукополисахаридоза из группы лизосомных болезней накопления, биохимически связанная с дефицитом фермента лизосом N-Ацетилгексозамин-4-сульфатсульфатазы.
GM2-ганглиозидо́з — тяжёлое наследственное заболевание, развивающееся в результате дефицита или недостаточной активности фермента гексозаминидазы и накопления в клетках ганглиозидов. Относится к лизосомным болезням накопления и имеет три варианта, связанные с мутациями в разных генах, которые оказывают вляние на активность общей гексозаминидазы. Два варианта заболевания больше известны под индивидуальными именами, полученными в честь авторов, впервые описавших их клиническую картину: болезнь Тея — Сакса, болезнь Сандхоффа. Третий вариант этой болезни носит название GM2 ганглиозидоз, вариант АБ. Болезнь Тея — Сакса вызвана мутацией в гене HEXA, кодирующий альфа-субъединицу гексоаминидазы А. Болезнь Сандхоффа вызвана мутацией в гене HEXB, кодирующий бета-субъединицу гексоаминидаз А и Б. GM2 ганглиозидоз, АБ вариант связан с нарушением в гене GM2, кодирующий белок-активатор GM2A.
GM2 ганглиозидо́з, вариа́нт АБ (англ. GM2-gangliosidosis, AB variant) — редкое аутосомно-рецессивное нарушение обмена веществ, связанное с мутацией гена GM2A. Характеризуется нормальной активностью β-гексозаминидаз А и Б и обусловлено недостаточностью активатора (белкового кофактора), необходимого для реализации ферментной активности в отношении субстрата. Заболевание клинически проявляется прогрессирующим разрушением нервных клеток головного и спинного мозга.

Аутосо́мно-рецесси́вное насле́дование — свойственный диплоидным эукариотам тип наследования признака, контролируемого рецессивными аллелями аутосомного гена. Для проявления мутации или болезни с таким типом наследования мутантный аллель, локализованный в аутосоме, должен быть унаследован от обоих родителей. Иными словами, мутация проявляется только в гомозиготном состоянии, то есть тогда, когда обе копии гена, расположенные на гомологичных аутосомах, являются повреждёнными. Если мутация находится в гетерозиготном состоянии, и мутантному аллелю сопутствует нормальный функциональный аллель, то аутосомно-рецессивная мутация не проявляется.
Синдро́м Ло́уренса-Му́на. Редкое аутосомно-рецессивное генетическое заболевание, характеризующиеся ожирением, умственной отсталостью, атаксией, дистрофией сетчатки, гипопитуитаризмом, большим количеством пальцев рук или ног. Синдром связан с нарушением функции гипоталамических центров.