
Синдром Лёша — Нихена — наследственное заболевание, характеризующееся увеличением синтеза мочевой кислоты и вызванное дефектом фермента гипоксантин-гуанинфосфорибозилтрансферазы, который катализирует реутилизацию гуанина и гипоксантина — в результате образуется большее количество ксантина и, следовательно, мочевой кислоты. Частота проявления синдрома 1 к 380000 живорожденных.

Синдро́м Ма́ртина — Белл — наследственное заболевание.
Мегалокорнеа — исключительно редкое непрогрессирующее врожденное увеличение роговицы, диаметр которой достигает и превышает 13 мм. Отмечается у некоторых пациентов с синдромом Марфана. Предполагается наличие двух подтипов мегалокорнеа: один наследуется по аутосомному пути, другой связан с Х-хромосомой. Последний подтип встречается чаще. Около 90 % случаев мегалокорнеа отмечается у лиц мужского пола.

Хлоридный канал 5 (англ. chloride channel 5, CLC-5, CLCN5) — потенциал-зависимый хлоридный канал, из суперсемейства CLCN. Канал обменивает ионы хлора Cl− на ионы водорода H+, то есть является антипортером. У человека он кодируется геном CLCN5, расположенным в X хромосоме.
Анеуплоиди́я — изменение кариотипа, при котором число хромосом в клетках не кратно гаплоидному набору (n). Отсутствие в хромосомном наборе диплоидного организма одной хромосомы называется моносомией (2n-1); отсутствие двух гомологичных хромосом — нуллисомией (2n-2); наличие дополнительной хромосомы называется трисомией (2n+1). Анеуплоидия возникает в результате нарушения сегрегации хромосом в митозе или мейозе. Анеуплоидия вызывает у человека некоторые наследственные синдромы. Анеуплоидия по аутосомам нарушает нормальное эмбриональное развитие и является одной из основных причин спонтанных абортов. Анеуплоидия характерна для опухолевых клеток, особенно для клеток сóлидных опухолей. Патологический фенотип при анеуплоидии формируется из-за нарушения дозового баланса генов, при моносомии дополнительный негативный вклад оказывает гемизиготное состояние генов моносомной хромосомы.

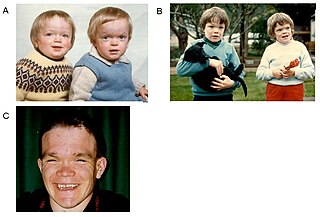
Синдром Рубинштейна-Тейби впервые описан в 1963 г. J. Rubinstein и Н. Taybi.
Популяционная частота — 1:25 000 — 30000.
Соотношение полов — M1:Ж1.
Тип наследования неизвестен.

Синдром Питта — Хопкинса — редкое генетическое заболевание, которое характеризуется умственной отсталостью, широким ртом и отличительными чертами лица, а также прерывистой гипервентиляцией с одышкой. По мере дальнейшего изучения синдрома Питта-Хопкинса спектр расстройств развития всё расширяется и может также включать проблемы с повышенной тревожностью, аутизмом, СДВГ и сенсорными расстройствами. Это связано с аномалией в хромосоме 18. В частности, это вызвано недостаточной экспрессией гена TCF4.

Pax6, или белок аниридии II типа, или окулоромбин — белок, тканеспецифический транскрипционный фактор. У человека закодирован в гене PAX6.

X-сцепленное рецессивное наследование — один из видов сцепленного с полом наследования. Такое наследование характерно для признаков, гены которых расположены на Х-хромосоме и которые проявляются только в гомозиготном или гемизиготном состоянии. Такой тип наследования имеет ряд врождённых наследственных заболеваний у человека, эти заболевания связаны с дефектом какого-либо из генов, расположенных на половой Х-хромосоме, и проявляются в случае, если нет другой Х-хромосомы с нормальной копией того же гена. В литературе встречается сокращение XR для обозначения X-сцепленного рецессивного наследования.
Альфа-маннозидо́з — редкое аутосомно-рецессивное наследственное заболевание из группы лизосомных болезней накопления, связанное с нарушением метаболизма олигосахаридов в результате снижения активности фермента лизосом альфа-маннозидазы. Также заболевание наносит ущерб в животноводстве.
Трихотиодистрофия (TTD) — аутосомно-рецессивное наследственное заболевание, характеризующееся ломким волосом и интеллектуальными нарушениями. TTD связана с диапазоном симптомов, затрагивающих органы эктодермы и нейроэктодермы. TTD могут быть подразделены на четыре синдрома: Примерно половина всех пациентов с трихотиодистрофией страдают светочувствительностью, которая делит классификацию синдромов с ней или без неё; BIDS и PBIDS, IBIDS и PIBIDS. Современное использование включает TTD-P (светочувствительная) и TTD.

Тройной синдром (ААА), также известный как синдром Ахалазия-Аддисонианизма-Алакримии или синдром Оллгрова , — редкий аутосомно-рецессивный врожденный порок. В большинстве случаев характеризуется отсутствием семейной истории о нём. Синдром был обнаружен Джереми Оллгроувом и его коллегами в 1978 году. ААА представляет аббревиатуру синдрома ахалазии-аддисонианизма-алакримии. Алакримия является обычно ранним проявлением. Это прогрессирующее заболевание, которое может длиться годами, чтобы развить полноценную клиническую картину.

Аладин (англ. Aladin), также известный как адракалин — белок, кодируемый у человека геном AAAS (англ. Aladin).
Синдром WAGR – это редкий наследственный аутосомно-доминантный синдром (частота 1:500000—1:1000000 человек, который клинически характеризуется сочетанием опухоли Вильмса, аниридии, урогенитальных аномалий и/или гонадобластомы и задержки психомоторного развития. Синдром встречается примерно в 8 % случаев врожденной аниридии, и в 0,75% всех случаев опухоли Вильмса.

Капикуа — белок человека, способный подавлять экспрессию ряда генов. Капикуа кодируется геном CIC, расположенным на длинном плече 19 хромосомы.
Однородительская дисомия — это явление, которое происходит, когда человек получает две копии хромосомы или части хромосомы от одного родителя и ни одной копии от другого родителя. Однородительская дисомия (ОД) может быть результатом гетеродисомии, при которой пара неидентичных хромосом наследуется от одного родителя, или изодисомии, при которой дублируется одна хромосома от одного родителя. Однородительская дисомия может иметь клиническое значение по нескольким причинам. Например, изодисомия или гетеродисомия могут нарушить специфичный для родителей геномный импринтинг, что приведет к нарушениям импринтинга. Кроме того, изодисомия приводит к большим блокам гомозиготности, что может привести к обнаружению рецессивных генов, аналогичное явление наблюдается у инбредных детей единокровных партнёров. Было обнаружено, что ОД встречается примерно в 1 из 2000 родов.
DDX3X-синдром — генетическое заболевание, развивающееся преимущественно у лиц женского пола. У пациентов наблюдается задержка умственного развития, признаки аутизма, СДВГ, сниженный мышечный тонус. Причина развития синдрома — мутации, затрагивающие ген DDX3X, расположенный на X-хромосоме. Клиническая картина зависит от характера конкретной мутации.
Пиридоксин-зависимая эпилепсия - тяжелая форма эпилепсии, при которой приступы с трудом поддаются контролю обычными противоэпилептическими препаратами. Заболевание развивается из-за мутаций гена ALDH7A1, приводящих к ускоренной деактивации активной формы витамина B6 в организме. Как правило, приступы начинаются вскоре после рождения, реже - через несколько месяцев, крайне редко - через несколько лет. Раннее назначение больших доз витамина B6 позволяет купировать приступы.
Синдром Хельсмуртел-ван дер Аа (ADNP-синдром) — редкое расстройство нервно-психического развития, при котором у пациентов отмечается умственная отсталость, признаки расстройств аутистического спектра, деформация черт лица (дисморфизм), мышечная гипотония, врожденные заболевания сердца, нарушения зрения, нарушения работы желудочно-кишечного тракта.
Синдром гиперфосфатазии с умственной отсталостью – заболевание, при котором у пациентов отмечается умственная отсталость различной степени тяжести и устойчивое повышение активности щелочной фосфатазы в крови. По состоянию на 2020 год было известно шесть генов, мутации которых приводят к развитию синдрома Мабри.
