
Альбинизм — наследственное заболевание, полное или почти полное отсутствие пигмента меланина или хлорофилла. Проявляется отсутствием нормальной вида окраски кожи, волос, шерсти, радужной и пигментной оболочек глаз, зелёных частей растений.

Синдром (болезнь) Марфана аутосомно-доминантное заболевание из группы наследственных патологий соединительной ткани. Синдром вызван мутациями генов, кодирующих синтез гликопротеина фибриллина-1, и является плейотропным. Заболевание характеризуется различной пенетрантностью и экспрессивностью. В классических случаях лица с синдромом Марфана высоки (долихостеномелия), имеют удлинённые конечности, вытянутые пальцы (арахнодактилия) и недоразвитие жировой клетчатки. Помимо характерных изменений в органах опорно-двигательного аппарата, наблюдается патология в органах зрения и сердечно-сосудистой системы, что в классических вариантах составляет триаду Марфана.

Синдром Холт — Орама, синдром сердце-рука — наследственное генетическое заболевание, поражающее развитие верхних конечностей и сердца. Обусловлен дефектом гена TBX5. Частота встречаемости — 1:100 000, 60 % случаев семейные, самцы и самки одинаково подвержены синдрому.

Синдром Ангельмана — обусловленная генетической аномалией патология, характеризующаяся такими признаками, как задержка психического развития, нарушения сна, припадки, хаотические движения, частый смех или улыбки. Также эту болезнь называют «синдром Петрушки» или «синдром счастливой куклы».

Синдро́м Ма́ртина — Белл — наследственное заболевание.
Синдром Торга-Винчестера — синдром, впервые описанный в 1969 году; у носителей которого отмечается спектр проявлений, в том числе сниженный рост вследствие патологии костей и суставов, помутнения роговицы, грубые черты лица, подкожные узелковые утолщения, огрубение кожи, гипертрихоз. Отклонения вызываются разнообразными мутациями гена MMP2. Их разнообразие привело к тому, что они были описаны как три различных синдрома: синдром Торга, синдром Винчестера, NAO-синдром, и лишь в 2006 году объединены под новым общим именем.

Синдро́м Экарди́ — редкий генетический порок развития, характеризующийся частичным или полным отсутствием одной из основных структур мозга — мозолистого тела, нарушением структуры сетчатки и инфантильными спазмами. Предположительно, синдром Экарди вызывается дефектом в Х-хромосоме, так как до сих пор наблюдался лишь у девочек и мальчиков с синдромом Клайнфельтера. Симптомы обычно возникают у детей в возрасте до 5 месяцев.

Синдро́м Да́уна — хромосомная болезнь, чаще всего вызванная тем, что хромосомы 21-й пары представлены тремя копиями вместо нормальных двух. Существует ещё две формы этого синдрома: транслокация хромосомы 21 на другие хромосомы — 4 % случаев, и мозаичный вариант синдрома — 5 %.
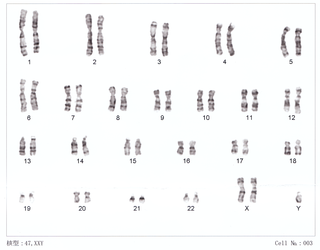
Синдром Клайнфельтера — генетическое отклонение. Клиническая картина синдрома описана в 1942 году в работах Гарри Клайнфельтера и Фуллера Олбрайта. Генетической особенностью этого синдрома является разнообразие цитогенетических вариантов и их сочетаний (мозаицизм). Обнаружено несколько типов полисомии по хромосомам X и Y у лиц мужского пола: 47, XXY; 47, XYY; 48, XXXY; 48, XYYY; 48 XXYY; 49 XXXXY; 49 XXXYY. Наиболее распространён синдром Клайнфельтера. Общая частота его колеблется в пределах 1 на 500—700 новорождённых мальчиков, что делает данный синдром первым по частоте встречаемости среди хромосомных болезней.

Синдро́м Шереше́вского — Тёрнера (СШТ) — хромосомный симптомокомплекс, обусловленный частичной или полной моносомией по X-хромосоме, в то время как здоровые женщины имеют и унаследованную от матери хромосому, и хромосому от отца. Характеризуется аномалиями физического развития, низкорослостью, половым инфантилизмом и другими разнящимися признаками. Традиционным методом верифицирования является кариотипирование. Терапия основана на приёме гормонов роста и эстрогенов.
Синдром Свайера, XY дисгенезия гонад, женская гонадальная дисгенезия или гонадальная дисгенезия — генетическое нарушение, вариант гипогонадизма с кариотипом 46,XY. Организм человека с синдромом Свайера имеет характерный для мужского организма набор хромосом, но половые железы представляют собой гонадный тяж и не производят гормоны. В результате он имеет женские гениталии, женскую репродуктивную систему и выглядит как женщина. В период полового созревания развитие вторичных половых признаков не происходит и наблюдается аменорея. Существует практика удаления гонад в раннем возрасте с целью предотвращения развития рака.

Синдро́м Нуна́н — редкая врождённая патология, как правило, наследуется по аутосомно-доминантному типу, носит семейный характер, однако встречаются и спорадически. Синдром предполагает наличие фенотипа, характерного для синдрома Шерешевского-Тернера у особей женского и мужского пола с нормальным генотипом.
Синдро́м де ля Шапе́ля — назван в честь исследователя, впервые охарактеризовавшего его в 1972 году. Синдром относится к редкой хромосомной патологии, возникающей в результате кроссинговера между X- и Y-хромосомами в процессе мейоза, в результате чего одна или обе X-хромосомы содержат нормальный мужской ген SRY. Распространённость данного синдрома составляет 4—5 на 100 000, что меньше встречаемости синдрома Клайнфельтера.

Синдром Питта — Хопкинса — редкое генетическое заболевание, которое характеризуется умственной отсталостью, широким ртом и отличительными чертами лица, а также прерывистой гипервентиляцией с одышкой. По мере дальнейшего изучения синдрома Питта-Хопкинса спектр расстройств развития всё расширяется и может также включать проблемы с повышенной тревожностью, аутизмом, СДВГ и сенсорными расстройствами. Это связано с аномалией в хромосоме 18. В частности, это вызвано недостаточной экспрессией гена TCF4.

Синдром Корнелии де Ланге — наследственное заболевание, проявляющееся умственной отсталостью и множественными аномалиями развития. Частота заболевания — примерно 1 на 10000.

Синдром Дуэйна — врождённый редкий тип косоглазия, чаще всего характеризуется неспособностью глаза двигаться наружу. Синдром был впервые описан офтальмологами Якобом Штиллингом (1887) и Зигмундом Тюрком (1896), а впоследствии стал носить имя Александра Дуэйна, который осветил этот синдром более подробно в 1905 году.
Синдро́м Ке́рнса — Се́йра (англ. Kearns–Sayre syndrome, сокращённо KSS) — митохондриальная миопатия с типичным началом до 20-летнего возраста. KSS является более серьёзным синдромным вариантом хронической прогрессирующей внешней офтальмоплегии, синдром, который характеризуется изолированным поражением мышц, контролирующих движения век и контролирующих движения глаз. Это приводит к птозу и офтальмоплегии соответственно. KSS включает в себя триаду уже описанных CPEO, двустороннюю пигментную ретинопатию и блокаду сердца. Другие области участия могут включать в себя мозжечковую атаксию, проксимальную мышечную слабость, глухоту, сахарный диабет, дефицит гормона роста, гипопаратиреоз или другие эндокринные нарушения. В обоих этих заболеваниях вовлечение мышц может начаться односторонним, но всегда развивается в двусторонний дефицит, который является прогрессирующим.
Синдром Коккейна , также называемый синдром Нил-Дингуолл — редкое аутосомно-рецессивное, нейродегенеративное расстройство, характеризующееся недостатком роста, нарушением развития нервной системы, аномальной чувствительностью к солнечному свету (фотосенсибилизация), заболеваниями глаз и преждевременным старением. Нездоровый вид и неврологические расстройства являются критериями для диагностики, а светочувствительность, нарушения слуха и ненормальные глаза — другие весьма общие черты. Возможны проблемы любого или всех внутренних органов. Это связано с группой расстройств, называемых лейкодистрофия. В основе расстройства лежит дефект механизма репарации ДНК. Интересно, в отличие от других дефектов репарации ДНК, пациенты с CS не предрасположены к раку или инфекции. Синдром Коккейна редок, но это разрушительная болезнь, которая, как правило, приводит к смерти в первом или втором десятилетии жизни. Мутация специфических генов в синдроме Коккейна известна, но широко распространенные эффекты и его отношения с репарацией ДНК еще не очень хорошо исследованы.
XX-дисгенезия гонад — тип женского гипогонадизма, при котором отсутствуют функциональные яичники, которые должны были бы вызвать половое созревание у нормальной в остальном девочки, с кариотипом 46, XX. Из-за отсутствия функциональных яичников у таких людей низкий уровень эстрогенов (гипоэстрогенизм) и высокий уровень ФСГ и ЛГ. Обычно при таком диагнозе назначают гормональную терапию эстрогенами и прогестероном.
Синдром Смит — Магенис (СМС) — наследственное заболевание, имеет такие проявления, как умственная отсталость, аномалии лица, проблемы со сном и многочисленные поведенческие проблемы, в том числе и такие, как самоповреждение. Частота синдрома Смит — Магенис составляет 1 случай на 15 000-25 000 человек. Синдром Смит — Магенис, как правило, обусловлен микроделецией в коротком плече 17-й хромосомы (17p), и иногда его называют 17p-синдром.

